5 Proteomic Landscapes of CCOC
root.dir <- rprojroot::find_rstudio_root_file()
knitr::opts_chunk$set(echo = TRUE)
knitr::opts_knit$set(root.dir = root.dir )
source(file.path(root.dir,'src/util.R'))
library(stringr)
library(dplyr)
library(plyr)
library(tidyr)
library(tibble)
library(ggplot2)
library(pheatmap)
library(heatmaply)
library(corrplot)
library(cowplot)
library(factoextra)
library(siggenes)
library(DT)5.1 Loading and Transforming RPPA data
Reverse Phase Protein Arrays (RPPA) is processed by Functional Proteomics RPPA Core at MD Anderson Cancer Center. A total of 66 samples (2 replicates for each cell line + medium) were probed with 453 antibodies. rppa_ab_info.csv contains antibody infromation and rppa_normal.csv contains 453 log2-transformed normalized relative protein level for each samples.
ab.info.path <- 'data/RPPA/rppa_ab_info.csv'
ab.info.df <- read.csv(ab.info.path)
ab.info.df$Gene.Name <- lapply(ab.info.df$Gene.Name,function(x){str_replace_all(x,"\\[PAR Modification\\]","PAR")})
rppa.normal.path <- 'data/RPPA/rppa_normal.csv'
#taking out ab_info and NormalLog2
rppa.normal.df <- read.csv(rppa.normal.path)
rppa.normal.df <- rppa.normal.df[,colSums(is.na(rppa.normal.df)) == 0]
ab.validation.status <- rppa.normal.df[4,-c(1:9)] %>% unlist %>% unname
rppa.normal.df <- rppa.normal.df[-c(1:8,10),]
names(rppa.normal.df) <- str_replace_all( rppa.normal.df[1,]," ","_" )
rppa.normal.df <- rppa.normal.df[-1,]
rppa.normal.df <- rppa.normal.df[!apply(is.na(rppa.normal.df) | rppa.normal.df == "", 1, all),]
rppa.normal.df <- rppa.normal.df[,colSums(rppa.normal.df != "") != 0 ]
cell.culture.type <- unname(sapply( rppa.normal.df["Sample_description"], function(x) str_extract(x,"(3D)|(2D)")))
cell.line <- sapply(rppa.normal.df$Sample_description, function(x){ y <- str_replace_all(x,"_[2-3]D_[1-2]$","")})
rppa.normal.df$Sample <- unname(sapply( rppa.normal.df["Sample_description"], function(x){ y <- str_replace_all(x,"_[1-2]$",""); str_replace_all(y,"-","_") }))
rppa.normal.df <- cbind( cell.line=cell.line, cell.culture.type=cell.culture.type , rppa.normal.df)
rppa.normal.df <- rppa.normal.df %>% filter(!str_detect(Sample_description,"NOSE16"))
#names(rppa.normal.df)<- c(names(rppa.normal.df)[c(1:9)], as.character(ab.name))
#names(rppa.normal.df)<- lapply(names(rppa.normal.df),function(x){str_replace_all(x,"\\[PAR Modification\\]","PARP")})
rownames(rppa.normal.df) <- str_replace_all(rownames(rppa.normal.df),"-","_")5.2 Removing Samples from RPPA that failed QC
The following samples are excluded for further as they do not meet QC threshold set by MD Anderson RPPA core for having an overall low protein concentration:
HCH1_3D_2
JHOC9_3D_1
JHOC9_3D_2
OVTOKO_3D_1
5.3 RPPA Experiment Summary
DT::datatable(ab.info.df %>% dplyr::count( Gene.Name,sort=TRUE),caption="Antibodies per gene count")ab.phosphorylation.count <-sum(str_detect(ab.info.df$Antibody.Name,"_p"))
protein.count <- ab.info.df$Gene.Name %>% unlist %>% unique %>% length
ab.count <- ab.info.df$Gene.Name %>% unlist %>% length
str_glue("In this assay, **{ab.count}** probes were available to capture **{protein.count}** proteins in which **{ab.phosphorylation.count}** is phosphorylated")In this assay, 453 probes were available to capture 392 proteins in which 91 is phosphorylated
5.4 Checking Technical Replicate Qulatity
5.4.1 Correlation plot
rppa.normal.df["Sample"] <- sapply( rppa.normal.df["Sample_description"], function(x){ y <- str_replace_all(x,"_[1-2]$",""); str_replace_all(y,"-","_") })
#calculating corrleation
M <- t(rppa.normal.df[,-c(1:10)] %>% dplyr::mutate_all(as.numeric))
colnames(M) <- rppa.normal.df$Sample_description
#colnames(M) <- sapply(colnames(M), function(x){ y <- str_replace_all(x," [0-9]$","")})
corrplot(cor(M,method = "spearman"), # Correlation matrix
method = "square", # Correlation plot method
type = "full", # Correlation plot style (also "upper" and "lower")
diag = TRUE, # If TRUE (default), adds the diagonal
tl.col = "black", # Labels color
bg = "white", # Background color
order = "hclust",
#hclust.method= "centroid",
hclust.method= "ward.D2",
tl.cex = 0.6,
col = NULL) # Color palette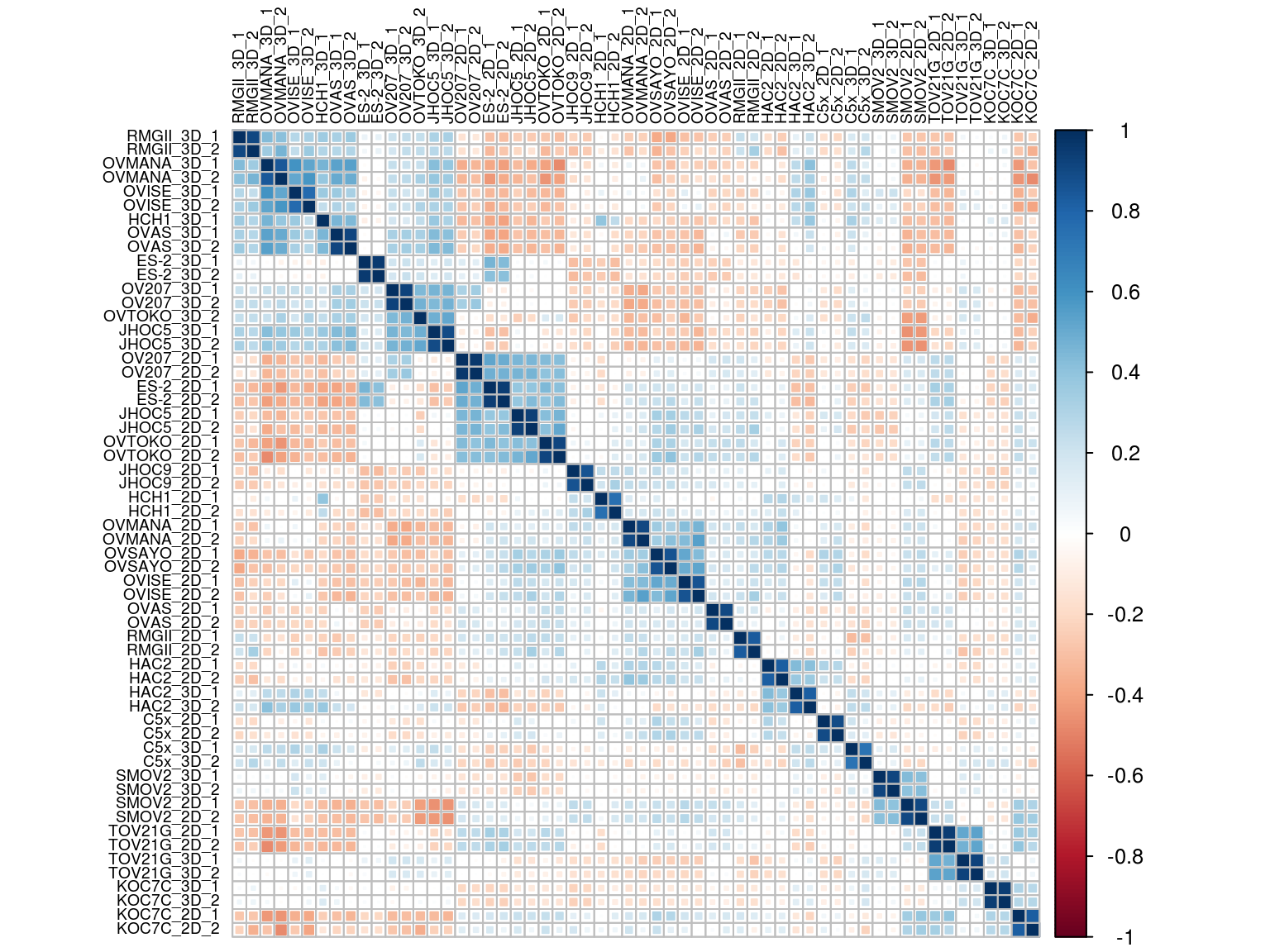
5.4.2 Scatter plot of protein level for each sample replicate
#get.pair <- function(x){ rppa.normal.df %>% filter(!! rlang::parse_expr(x)) %>% dplyr::select(Sample_description) }
#rppa.group <- sapply(unique(rppa.normal.df["Sample"]),function(x){ expr <- paste0('Sample == "', x , '"' ) ; get.pair(expr) })
rppa.group <- split(rppa.normal.df[,-c(1:5,7:10)] %>% dplyr::mutate_at(vars(-c("Sample")),as.numeric), f =rppa.normal.df$Sample)
rppa.group.plot <- lapply(rppa.group, function(df){ if(dim(df)[[1]] >1){
col.name <-rownames(df);
df <-data.frame(t(df)[-1,])%>% mutate_all(as.numeric)
ggplot(df,aes(x= .data[[ col.name[[1]] ]], y= .data[[ col.name[[2]] ]] ))+ geom_point() + theme_minimal()+geom_abline()}})
rppa.group.plot <- rppa.group.plot[lengths(rppa.group.plot) != 0]
plot_grid(plotlist = rppa.group.plot, align = "h")
5.4.3 Heatmap for all sample replicates
heat.map.color <- viridis(n = 256, alpha = 1, begin = 0, end = 1, option = "viridis")
ann.colors <- list(cell.culture.type= c(`3D`= "#1B9E77",`2D` = "#D95F02"))df <- as.data.frame(rppa.normal.df[c("cell.culture.type","cell.line")])
rownames(df) <- rppa.normal.df$Sample_description
select <- order( apply(M, 1, sd) ,
decreasing=TRUE)
M <- apply(M,2,function(x){(x - mean(x)) / sd(x)})
pheatmap(M[select,] , cluster_rows=FALSE, show_rownames=FALSE, color=heat.map.color,
cluster_cols=TRUE, annotation_col=df, fontsize = 8,annotation_colors=ann.colors, clustering_method= "ward.D2")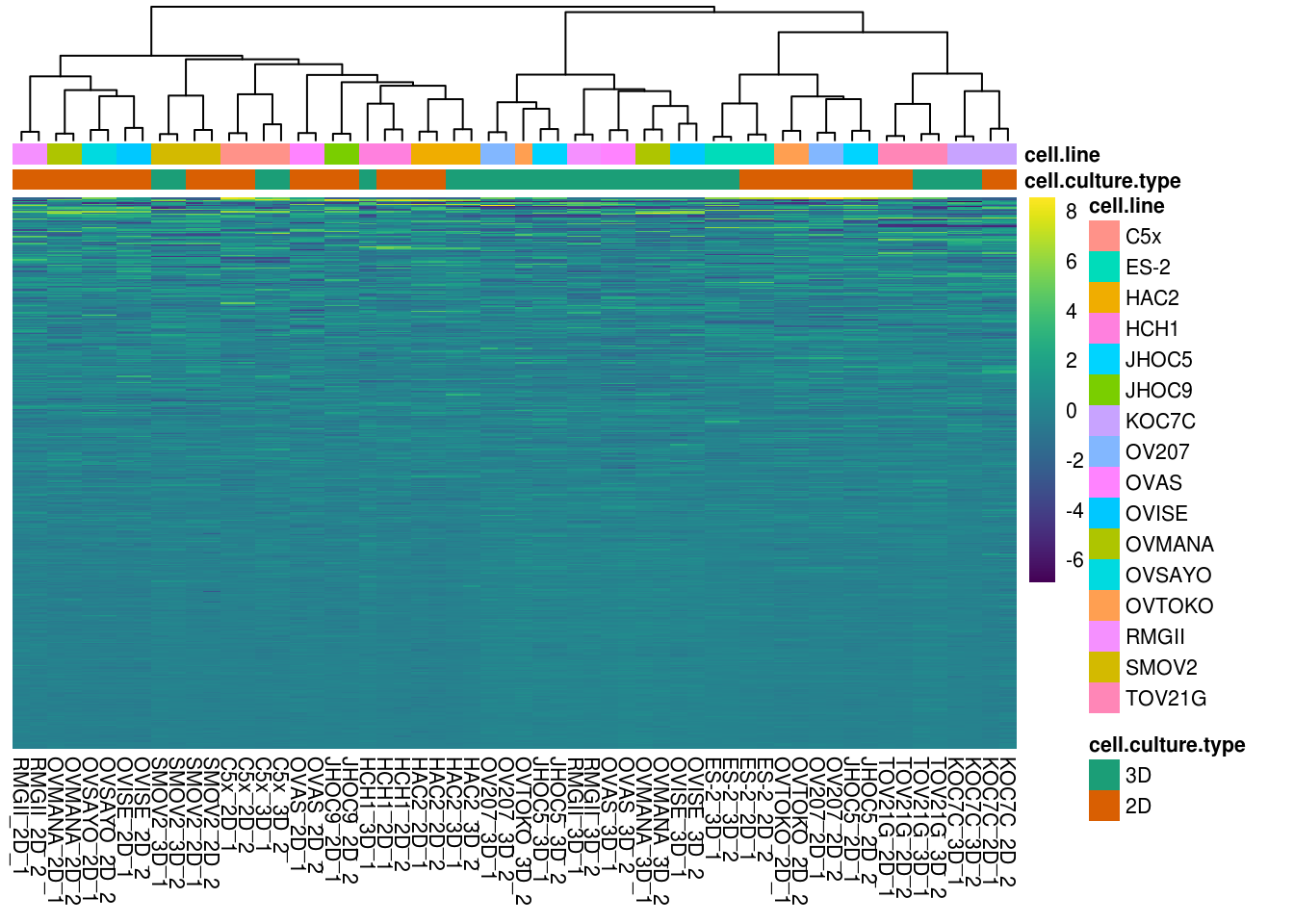
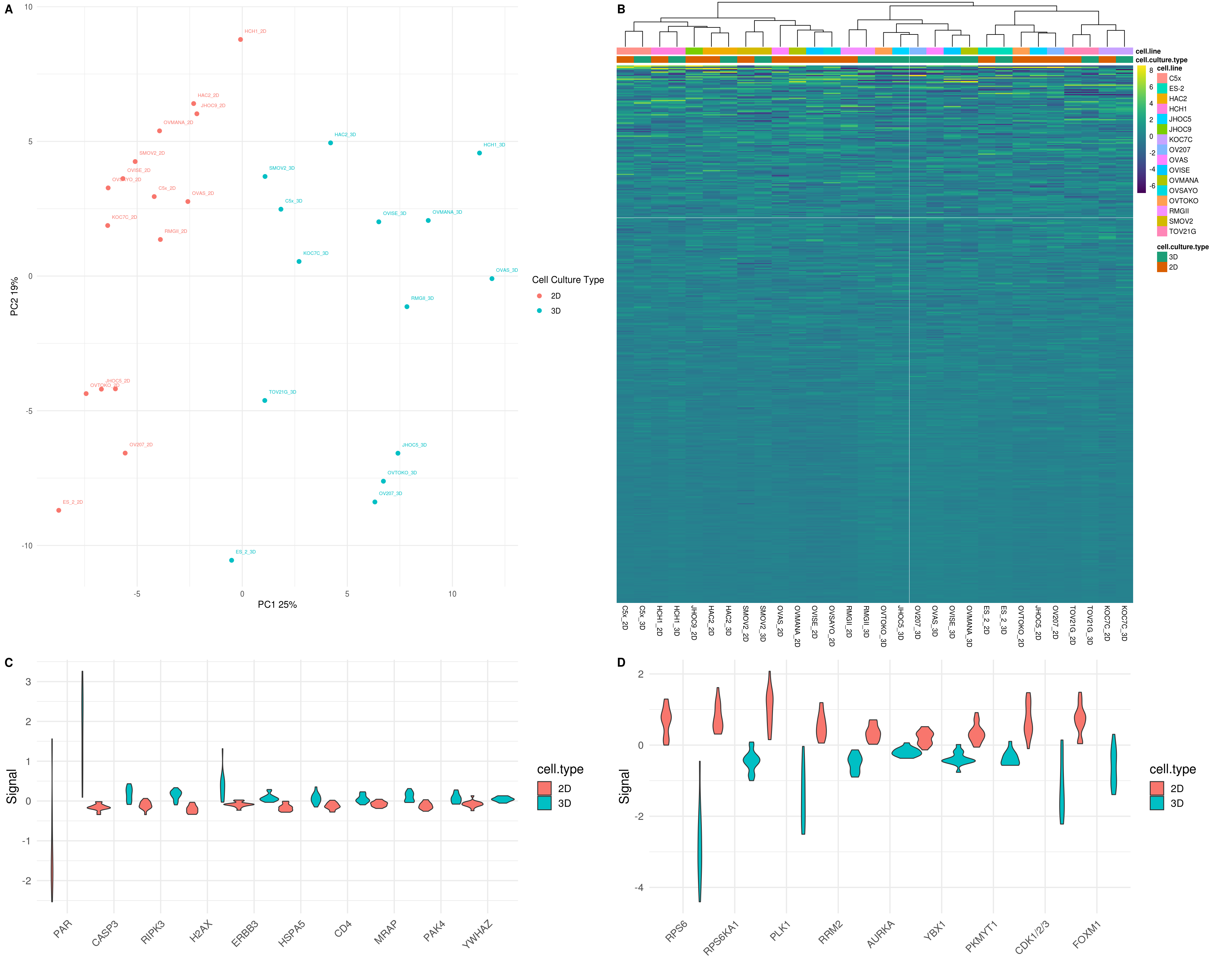
5.4.4 PCA plot all sample replicates
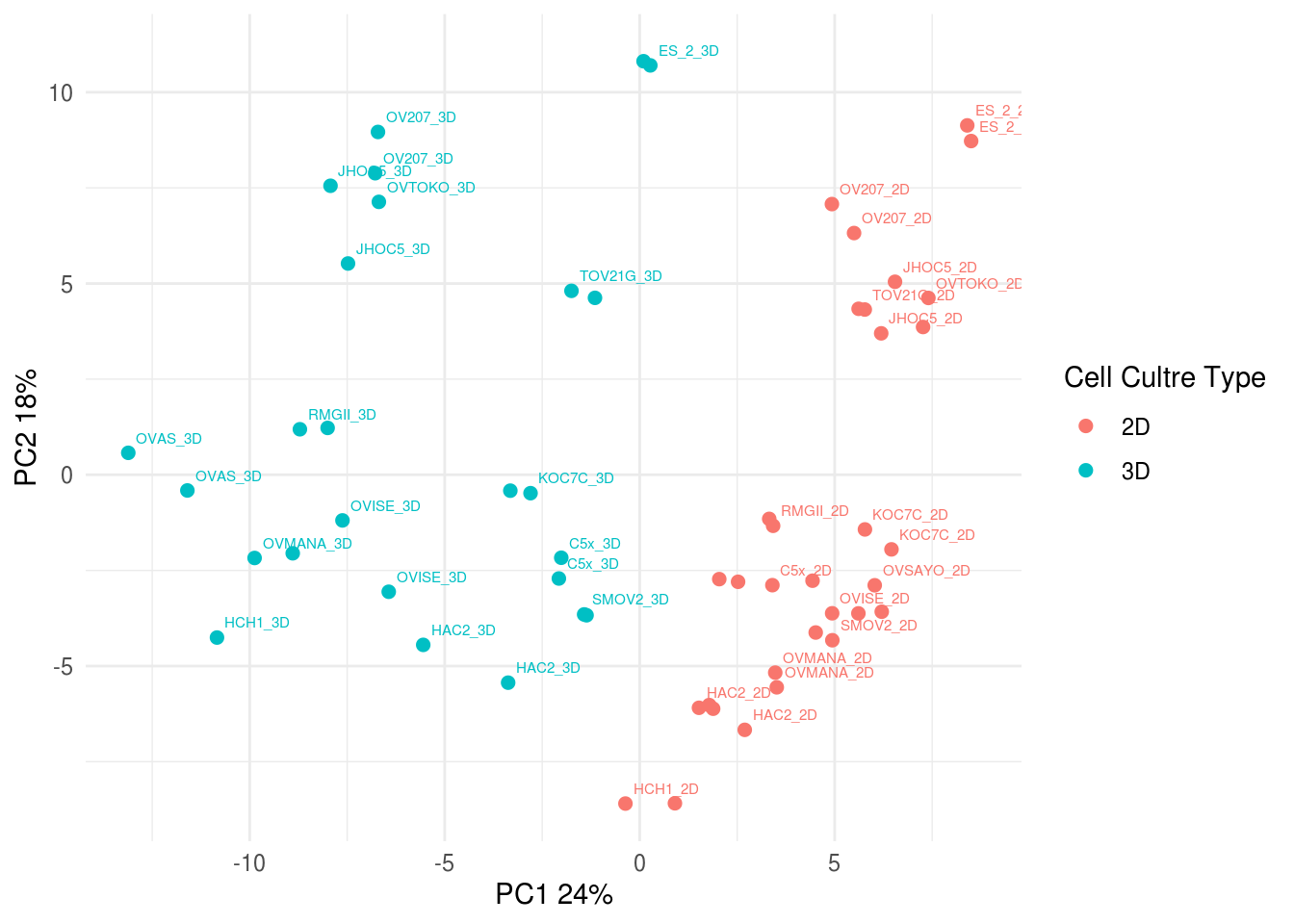
5.5 Merging replicates
To conduct differenial protein expression analysis, replicates for each samples were merged by taking the mean of log2-transformed normalized relative protein level. PCA and Heatmap was carried out to confirm the merged samples were similar to individual replicates.
rppa.normal.df.combined <- rppa.normal.df[,-c(3:5,7:10)] %>% group_by(Sample,cell.culture.type,cell.line) %>% mutate_at(vars(-c("Sample","cell.line","cell.culture.type")),as.numeric) %>% summarise_if(.,is.numeric,"mean")
rownames(rppa.normal.df.combined) <-rppa.normal.df.combined$Sample## Warning: Setting row names on a tibble is deprecated.5.5.1 Heatmap for merged RPPA data
M <- t(rppa.normal.df.combined %>% as.data.frame %>%dplyr::select(!c(Sample,cell.culture.type,cell.line)))
colnames(M) <- rppa.normal.df.combined$Sample
#geting metadata from rppa table
df <- as.data.frame(rppa.normal.df[c("cell.culture.type","cell.line")])
rownames(df) <- rppa.normal.df$Sample_description
df<- df %>% unique
rownames(df) <- sapply(rownames(df), function(x){ y <- str_replace_all(x,"_[1-2]$",""); z<- str_replace_all(y,"-","_") ; return(z)})
#rppa.normal.df.combined.heatmap <- merge(rppa.normal.df.combined, df, by.x="Sample", by.y="row.names")
select <- order( apply(M, 1, sd) ,
decreasing=TRUE)
M <- apply(M,2,function(x){(x - mean(x)) / sd(x)})
heat.map<- pheatmap(M[select,] , cluster_rows=FALSE, show_rownames=FALSE,color=heat.map.color,
cluster_cols=TRUE, annotation_col=df, fontsize = 8,annotation_colors=ann.colors, clustering_method= "ward.D2")
heat.map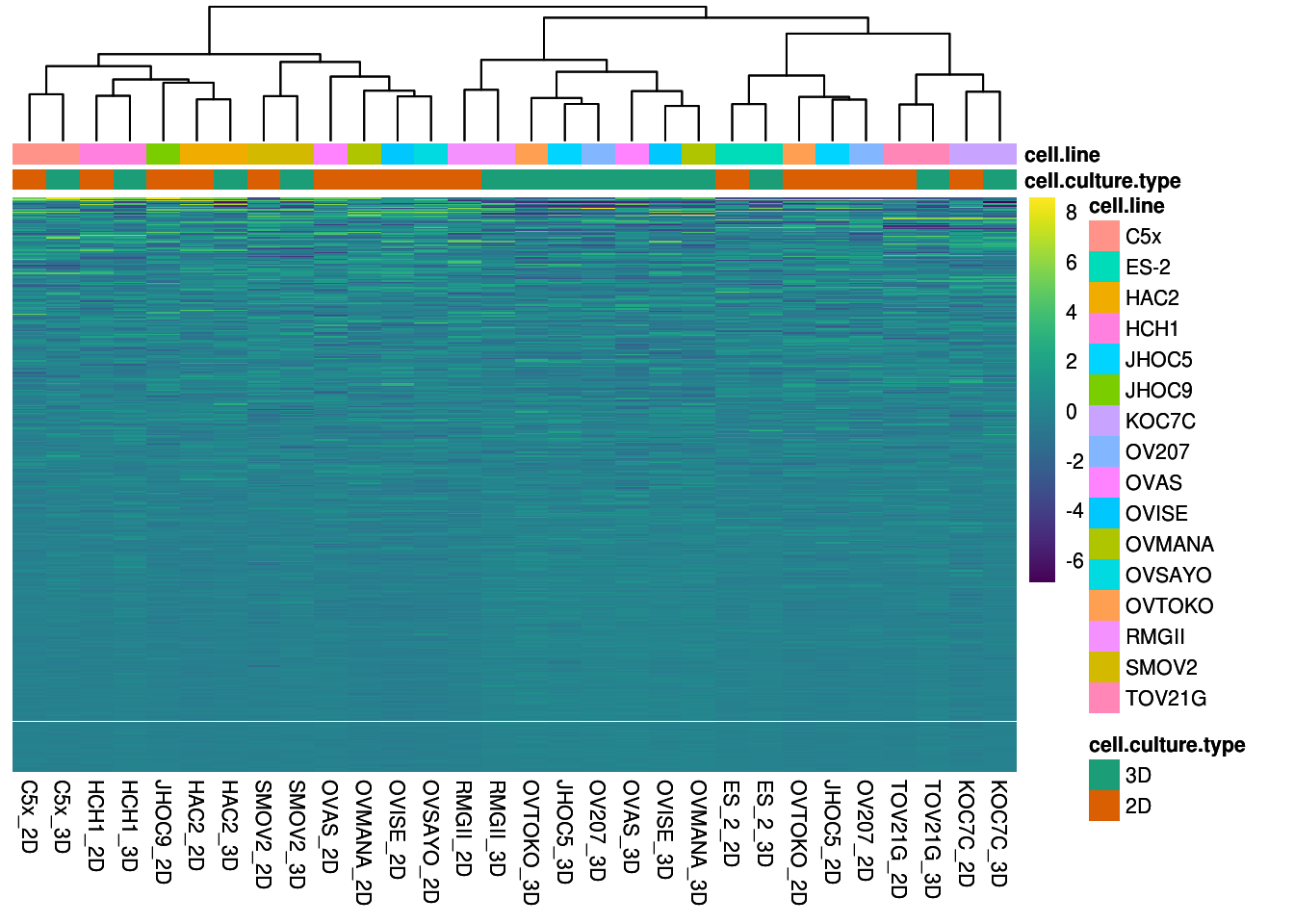
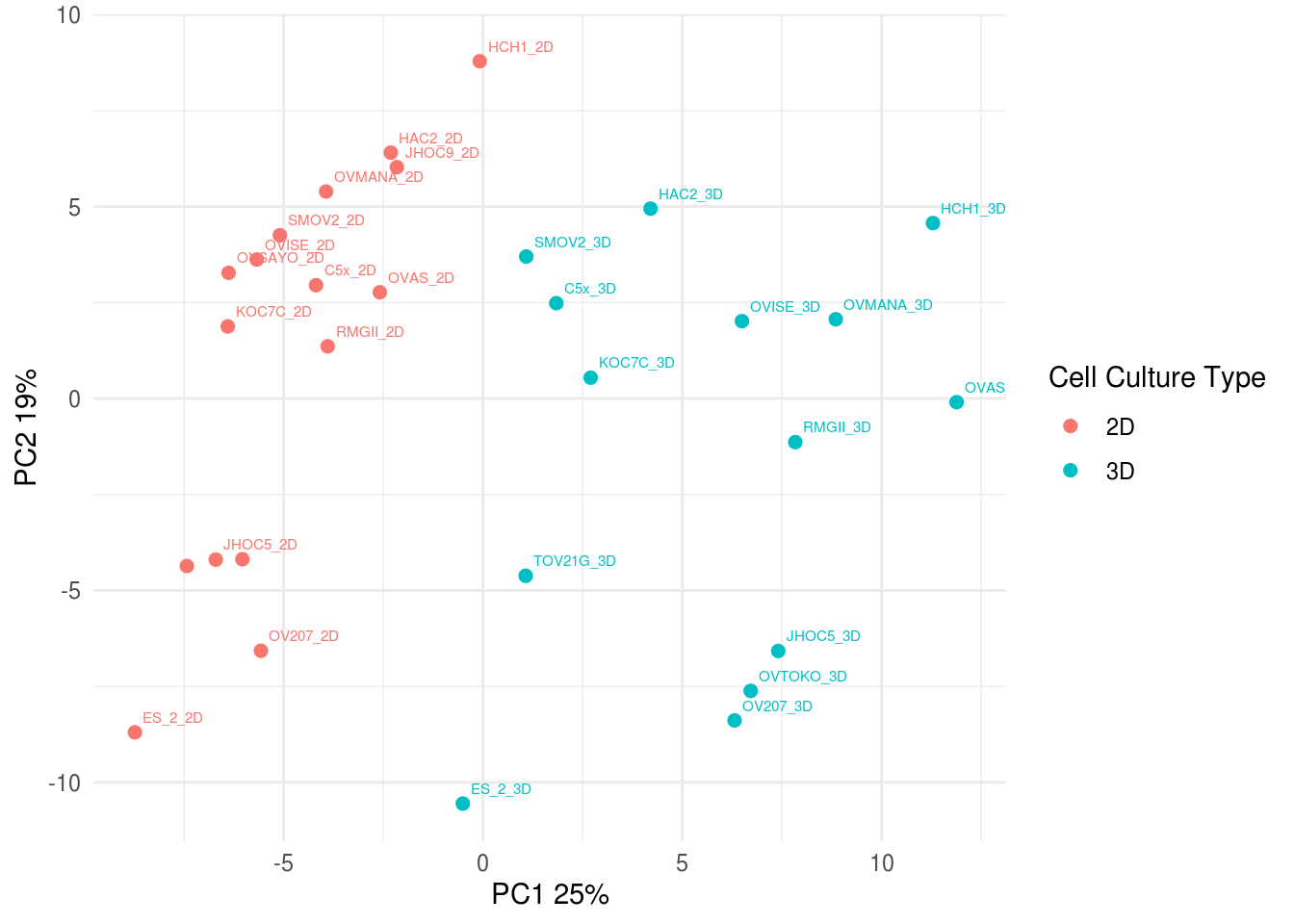
5.5.2 PCA for merged RPPA data
## Warning: Setting row names on a tibble is deprecated.
5.6 Significance Analysis of Microarray (SAM)
5.7 SAM Results
5.7.1 plot
sam.result <- sam( t(pca.tbl), sapply(rppa.normal.df.combined$cell.culture.type, function(x) { if (x == "2D") 0 else 1} ), method=d.stat, gene.names = names(pca.tbl))
plot(sam.result) ## To obtain a SAM plot, delta has to be specified in plot(object,delta,...).
5.7.2 table
| Delta | p0 | False | Called | FDR | cutlow | cutup | j2 | j1 |
|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.4159848 | 371.17 | 420 | 0.3676217 | -0.2962185 | 0.1596744 | 199 | 232 |
| 1.1 | 0.4159848 | 32.23 | 214 | 0.0626504 | -1.9398355 | 1.7717970 | 97 | 336 |
| 2.1 | 0.4159848 | 0.94 | 108 | 0.0036206 | -3.4676973 | 3.1939353 | 45 | 390 |
| 3.2 | 0.4159848 | 0.01 | 45 | 0.0000924 | -4.8968783 | 5.3570293 | 38 | 446 |
| 4.2 | 0.4159848 | 0.00 | 27 | 0.0000000 | -5.9973094 | 7.3867023 | 26 | 452 |
| 5.2 | 0.4159848 | 0.00 | 18 | 0.0000000 | -7.0261086 | Inf | 18 | 453 |
| 6.2 | 0.4159848 | 0.00 | 8 | 0.0000000 | -8.3942860 | Inf | 8 | 453 |
| 7.2 | 0.4159848 | 0.00 | 5 | 0.0000000 | -9.6834131 | Inf | 5 | 453 |
| 8.2 | 0.4159848 | 0.00 | 2 | 0.0000000 | -11.9922925 | Inf | 2 | 453 |
| 9.3 | 0.4159848 | 0.00 | 0 | 0.0000000 | -Inf | Inf | 0 | 453 |
5.8 SAM Results for ∆ = 1.21
FDR threshold is set to be less than 5% which will make the value of parameter delta be somewhere between 1.009075 and 1.009076. Rounding it up to 1.21 for further analysis. NB The delta estimate method seems to be heuristic which might yield different delta in each runs that lead to different # of total DE protein. However, delta doesn’t affect top ranked DE proteins.
5.8.1 SAM plot
## The threshold seems to be at
## Delta Called FDR
## 5 1.207411 208 0.050158
## 6 1.207412 207 0.048994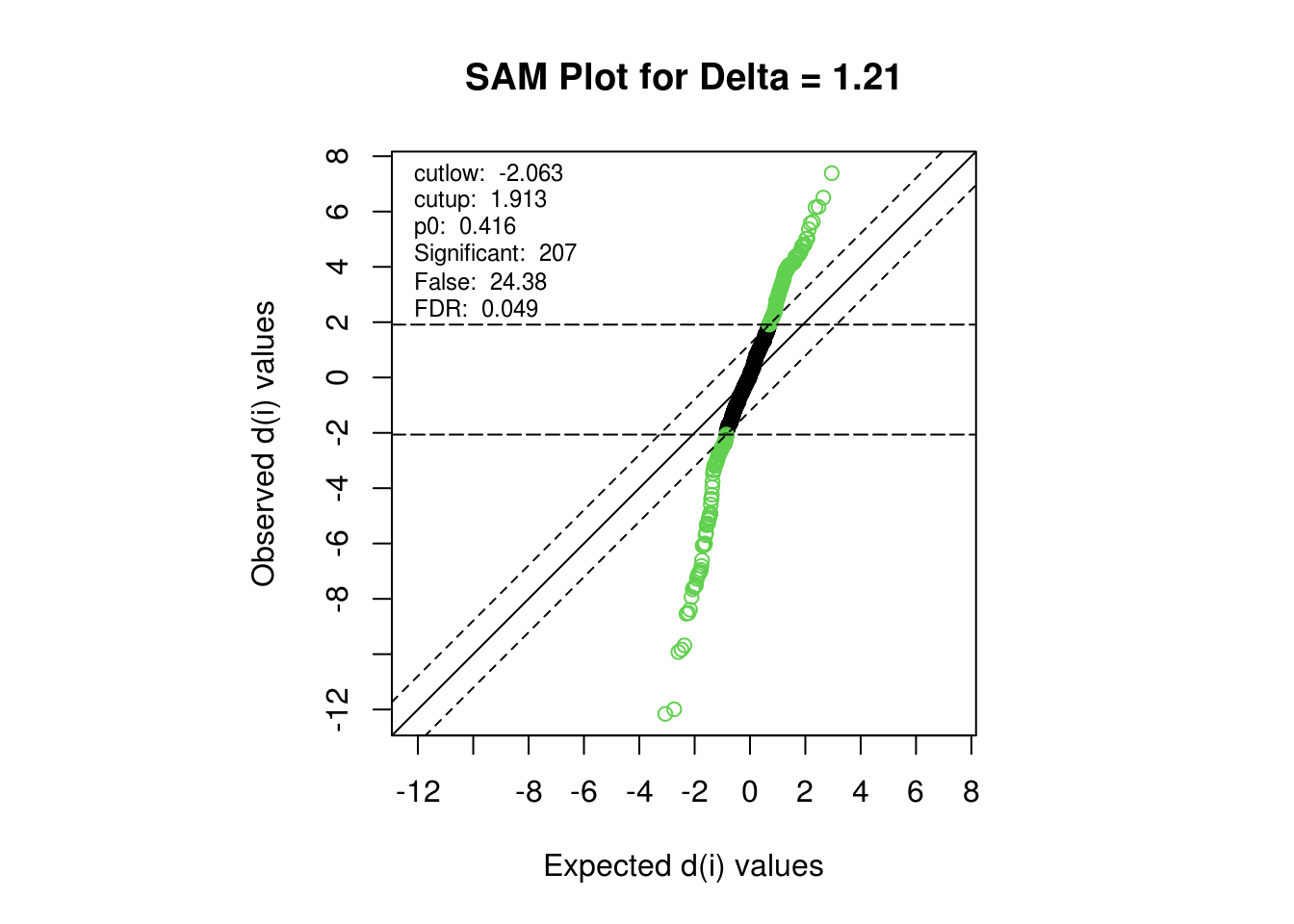
5.8.2 SAM summary
| Delta | p0 | False | Called | FDR | cutlow | cutup | j2 | j1 |
|---|---|---|---|---|---|---|---|---|
| 1.21 | 0.4159848 | 24.38 | 207 | 0.0489938 | -2.063406 | 1.913195 | 95 | 341 |
5.8.3 SAM protein table
gene.table <- sam.genes@mat.sig %>% mutate_if(is.numeric,round,4) %>% rownames_to_column("Antibody.Name.in.Heatmap") %>% mutate(gene.ab= factor(Antibody.Name.in.Heatmap,levels=ab.info.df$Antibody.Name.in.Heatmap, labels=ab.info.df$Gene.Name)) %>% left_join(ab.info.df[c("Antibody.Name.in.Heatmap","Validation.Status")],by="Antibody.Name.in.Heatmap") %>% dplyr::select(-c("Row"))
DT::datatable(gene.table,colnames = c("Ab","d.value","standard deviations","unadj p-val","q-val","Fold Change","Genes","validation status"),extensions =c('Buttons','Scroller'), options = list(
dom = 'Bfrtip',
buttons = c('copy', 'csv', 'excel', 'pdf', 'print'),
"scrollX"= TRUE
)) %>%
formatStyle(
'Antibody.Name.in.Heatmap',
`font-size` = '12px',
)5.9 Top 10 SAM up/down-regulated proteins for ∆ = 1.21
5.9.1 Boxplot for top 10 SAM up/down-regulated proteins for ∆ = 1.21
#remapping pca_tbl with gene name
names(pca.tbl) <- factor(names(pca.tbl),levels=ab.info.df$Antibody.Name.in.Heatmap, labels=ab.info.df$Gene.Name)## Warning: The `value` argument of `names<-` must be a character vector as of tibble 3.0.0.n <- 10
up.gene.idx <- sam.genes@mat.sig[order(sam.genes@mat.sig$d.value, decreasing=TRUE),]$Row[1:n]
down.gene.idx <- sam.genes@mat.sig[order(sam.genes@mat.sig$d.value),]$Row[1:n]
tbl.up <-data.frame(cell.type=rppa.normal.df.combined$cell.culture.type, stack(pca.tbl[up.gene.idx]))
up.plot<- ggplot(tbl.up, aes(x=ind, y=values, fill=cell.type))+ geom_violin(width=1.3) + xlab("") + ylab("Signal")+scale_x_discrete(guide = guide_axis(angle = 45))+theme_minimal(base_size = 15)
tbl.down <-data.frame(cell.type=rppa.normal.df.combined$cell.culture.type, stack(pca.tbl[down.gene.idx]))
down.plot <- ggplot(tbl.down, aes(x=ind, y=values, fill=cell.type))+ geom_violin(width=1.3) + xlab("") + ylab("Signal")+scale_x_discrete(guide = guide_axis(angle = 45))+ theme_minimal(base_size = 15)
plot_grid(plotlist = list(up.plot, down.plot))## Warning: position_dodge requires non-overlapping x intervals## Warning: position_dodge requires non-overlapping x intervals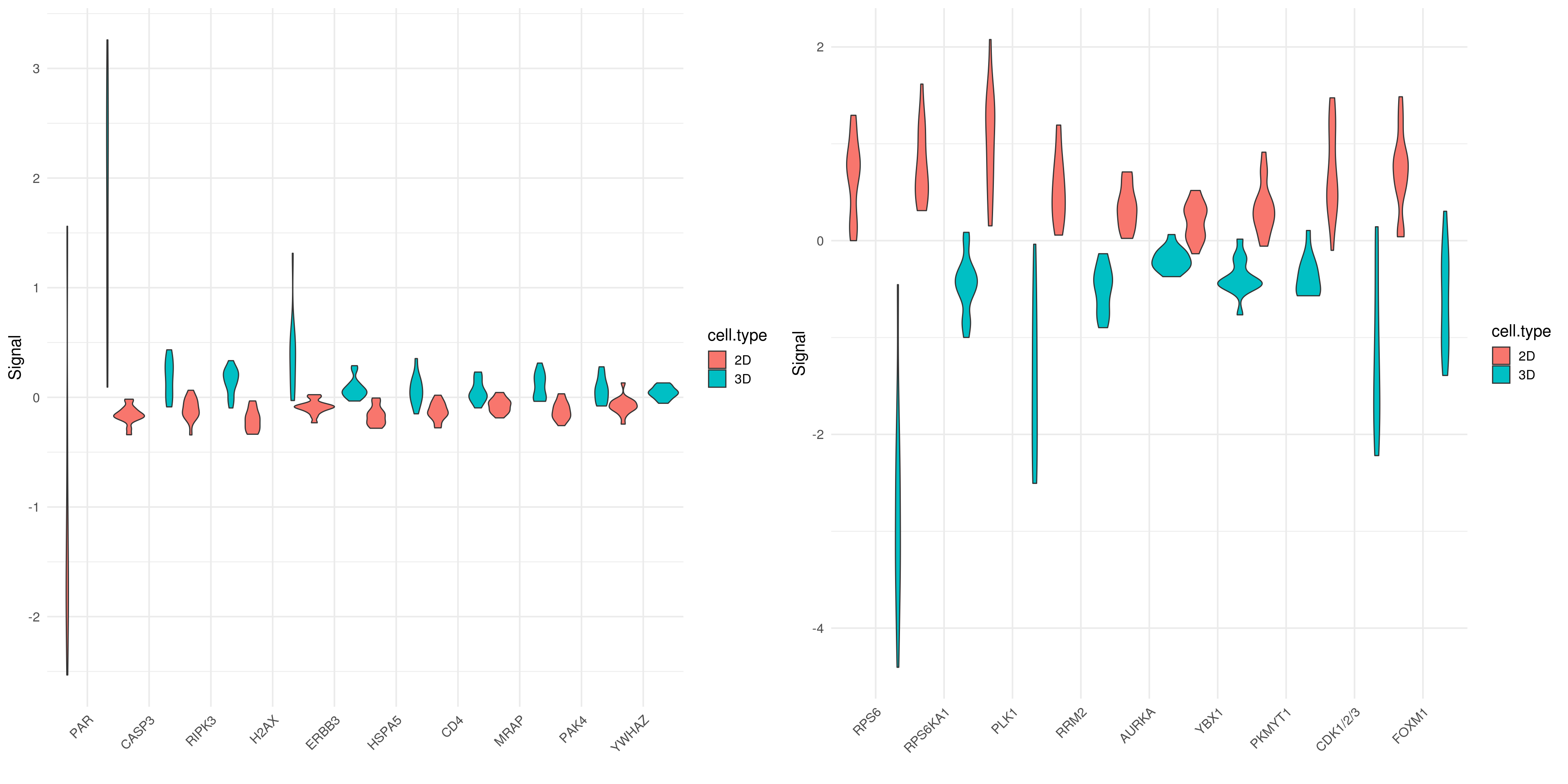
5.9.2 Heatmap for top 10 SAM up/down-regulated proteins for ∆ = 1.21
Subsetting the genes of ineterest from previous heatmap link text top 10 up-regulated protein doesn’t show too much varaince among each cell lines except PRAP (PAR-R-C). On the other hand,top 10 down-regulated proteins show strong differentiation
5.9.2.1 static heatmap
rownames(M) <- factor(rownames(M),levels=ab.info.df$Antibody.Name.in.Heatmap, labels=ab.info.df$Gene.Name)
up.heatmap <- pheatmap(M[up.gene.idx,] , cluster_rows=FALSE, color=heat.map.color ,
cluster_cols=FALSE, annotation_col=df, fontsize = 8,annotation_colors=ann.colors, clustering_method= "ward.D2",main="Up")
down.heatmap <- pheatmap(M[down.gene.idx,] , cluster_rows=FALSE, color=heat.map.color,
cluster_cols=FALSE, annotation_col=df, fontsize = 8,annotation_colors=ann.colors, clustering_method= "ward.D2", main="Down")

5.9.2.2 Interactive heatmap
#top 10 SAM up-regulated protein
heatmaply(M[up.gene.idx,], hclust_method = "ward.D2", main="Top 10 up-regulated proteins")#top 10 SAM down-regulated protein
heatmaply(M[down.gene.idx,], hclust_method = "ward.D2", main="Top 10 down-regulated proteins")## Warning in fix_not_all_unique(rownames(x)): Not all the values are unique - manually added prefix numbers
5.10 Figure 4. Targeted proteomic profiling of CCOC models
## Warning: position_dodge requires non-overlapping x intervals
## position_dodge requires non-overlapping x intervals