3 Transcriptomatic Landscapes of CCOC models
3.1 Data Preparation and QC
root.dir <- rprojroot::find_rstudio_root_file()
knitr::opts_chunk$set(echo = TRUE)
knitr::opts_knit$set(root.dir = root.dir )
source(file.path(root.dir,'src/util.R'))
library(DESeq2)
library(dplyr)
library(tidyr)
library(tibble)
library(stringr)
library(knitr)
library(purrr)
library(itertools)
library(cowplot)
library(pheatmap)
library(heatmaply)
library(vsn)
library(ggplot2)
library(ggrepel)
library(gridExtra)
library(corrplot)
library(M3C)
library(UpSetR)
library(kableExtra)
library(biomaRt)3.1.1 Loading gene expression data set
3.1.1.1 Loading count matrix
path.CCOC.RNA.count.table <- "data/RNA-seq/count_matrix/cell_line/KL-6799--04--27--2019_COUNTS.csv"
path.CCOC.RNA.count.table_2 <- "data/RNA-seq/count_matrix/cell_line/FA-9840--06--23--2020_COUNTS.csv"
tbls <- lapply(c(path.CCOC.RNA.count.table,path.CCOC.RNA.count.table_2),function(x){read.csv(file=x, row.names = 1,header = T, sep=",")} )3.1.1.2 Loading Cell line
#import cell line name
col.label.name.path <-'data/RNA-seq/CCOC_RNA_table_col.csv'
col.csv.tbl <- read.csv(file = col.label.name.path, header = T)
CCOC.RNA.count.table.col.name <- col.csv.tbl$KL.6799..04..27..2019_COUNTS %>% na_if("") %>% na.omit
CCOC.RNA.count.table_2.col.name <- col.csv.tbl$FA.9840..06..23..2020_COUNTS %>% na_if("") %>% na.omit
col.names <-list(CCOC.RNA.count.table.col.name,CCOC.RNA.count.table_2.col.name)
names(col.names)<- c(1,2)
tbls <- mapply(function(x,y){z<- paste(col.names[[y]],y);setNames(x,z) },tbls,names(col.names))3.1.1.3 Making DGE object
3.1.1.3.1 merging tables into a single table
3.1.1.3.2 Making colData
#building colData
#cell.name <- as.factor(sapply(names(CCOC.RNA.count.table), function(x){ y <- str_replace_all(x,"-[1-2]$","")}))
cell.name <- as.factor(names(CCOC.RNA.count.table))
cell.batch <- as.factor(do.call(c,sapply(as.list(enumerate(tbls)),function(x){rep(x[[1]],length(x[[2]]))})))
cell.type <- as.factor(sapply(names(CCOC.RNA.count.table), function(x){if(str_detect(x,"3D")){"3D";}else "2D"}))
cell.line <- as.factor(cell.name %>% str_trim() %>% str_replace(., "[2-3]D-","") %>% str_replace(.,"-[0-9] [0-9]$",""))
colData <- data.frame(type=cell.type,name=cell.name , cell.line=cell.line, cell.batch= cell.batch)
rownames(colData) <- names(CCOC.RNA.count.table)
colData$name <- str_replace_all(colData$name,"-[0-9] [0-9]$","") %>% as.factor3.1.2 Checking Technical Replicate Qulatity
msd.ntd <- meanSdPlot(assay(ntd), plot = F)
msd.vsd <- meanSdPlot(assay(vsd), plot = F)
# msd.rld <- meanSdPlot(assay(rld), plot = F)
ymax <- max(c(max(msd.ntd$gg$data$py),
max(msd.vsd$gg$data$py)))#,
#max(msd.rld$gg$data$py)))
p <- list()
p[[1]] <- msd.ntd$gg + ggtitle('log2(n + 1)') + ylim(c(0,ymax))
p[[2]] <- msd.vsd$gg + ggtitle('Variance stabilizing transformation') + ylim(c(0,ymax))
# p[[3]] <- msd.rld$gg + ggtitle('Regularized log transformation') + ylim(c(0,ymax))
library(cowplot)
plot_grid(plotlist = p, align = 'hv', ncol = 2)#3)## Warning: Removed 9 rows containing missing values (geom_hex).## Warning: Removed 12 rows containing missing values (geom_hex).
3.1.2.1 Correlation plot
M <- counts(dds)
colnames(M) <- sapply(colnames(M), function(x){ y <- str_replace_all(x," [0-9]$","")})
corrplot(cor(M), # Correlation matrix
method = "square", # Correlation plot method
type = "full", # Correlation plot style (also "upper" and "lower")
diag = TRUE, # If TRUE (default), adds the diagonal
tl.col = "black", # Labels color
bg = "white", # Background color
order = "hclust",
hclust.method= "ward.D2",
tl.cex = 0.8,
col = NULL) # Color palette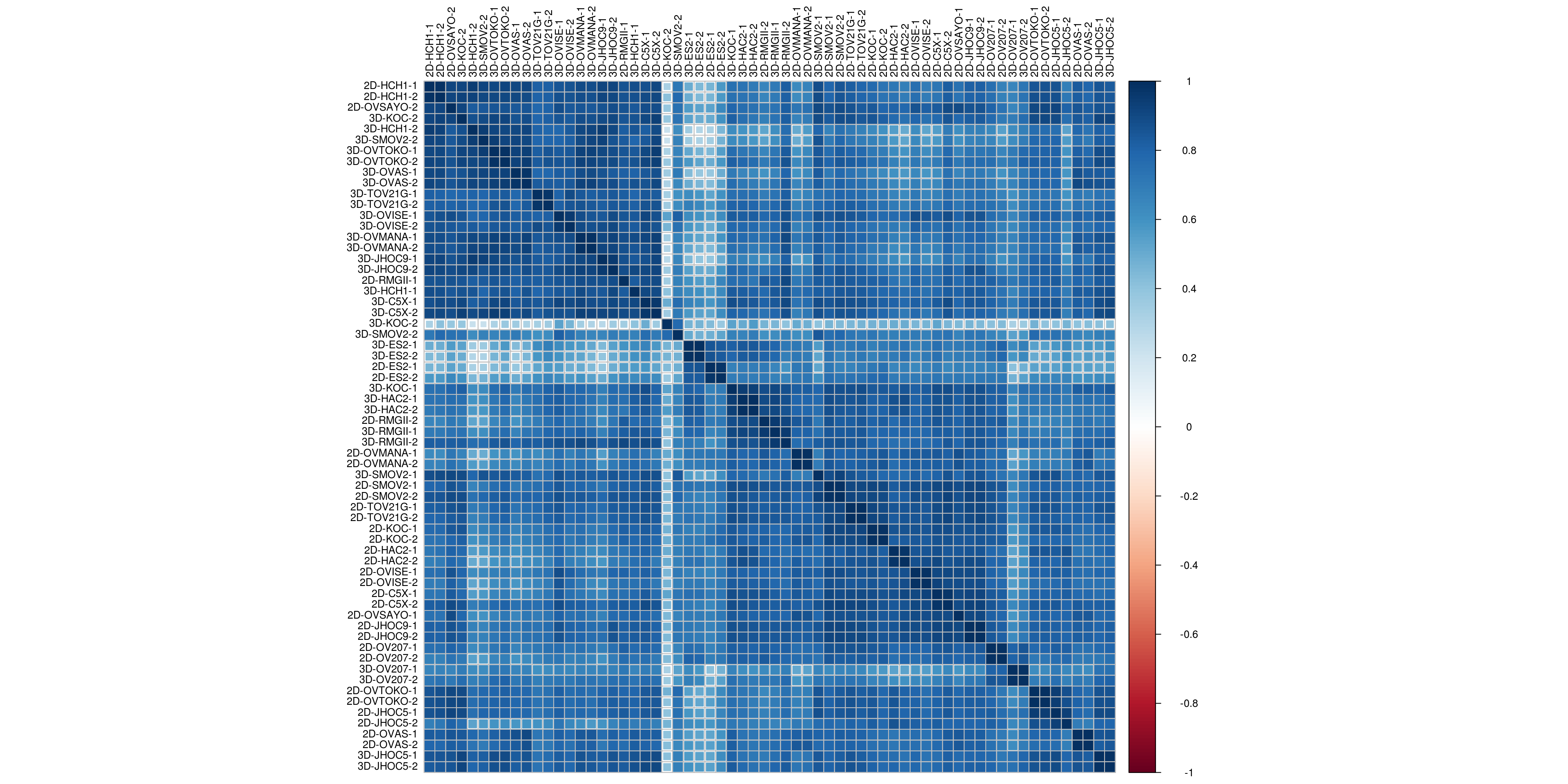
3.1.2.2 Sample to Sample Distance
sampleDists <- dist(t(assay(vsd)))
sampleDistMatrix <- sampleDists %>% as.matrix %>% as.data.frame
rownames(sampleDistMatrix) <- rownames(colData(vsd))
colnames(sampleDistMatrix) <- vsd$name
colors <- heat.map.color <- viridis(n = 256, alpha = 1, begin = 0, end = 1, option = "mako")
pheatmap(sampleDistMatrix,
clustering_distance_rows=sampleDists,
clustering_distance_cols=sampleDists,
clustering_method = 'ward.D2',
col=colors, fontsize = 10, fontsize_row = 4)3.1.2.3 PCA
show.labels <- TRUE # change to TRUE if you want to show the sample IDs
pcaData <- plotPCA.DESeqTransform(vsd, intgroup='type', returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
# adding cell line name into pcaData dataframe
pcaData <- merge(pcaData, unique(colData[c("name","cell.line")]) , by="name")
if(show.labels){
#label <- pcaData$cell.name
label <- pcaData$name
}else{
label <- NA
}
PC.list <-c(1:4)
g.plot <- lapply( PC.list , function(i){
x=paste("PC",i,sep="")
y=paste("PC",i+1,sep="")
return( ggplot(pcaData[c(x,y,"cell.line","type")], aes( .data[[x]], .data[[y]], color= cell.line ,shape=type ,label = label)) +
geom_point(size=2) +
xlab(paste0(x,": ",percentVar[i],"% variance")) +
ylab(paste0(y,": ",percentVar[i+1],"% variance")) +
geom_text_repel(show.legend = F,size=1)+
guides(colour="none"))+
theme_minimal()
})
plot_grid(plotlist = g.plot, align = "h")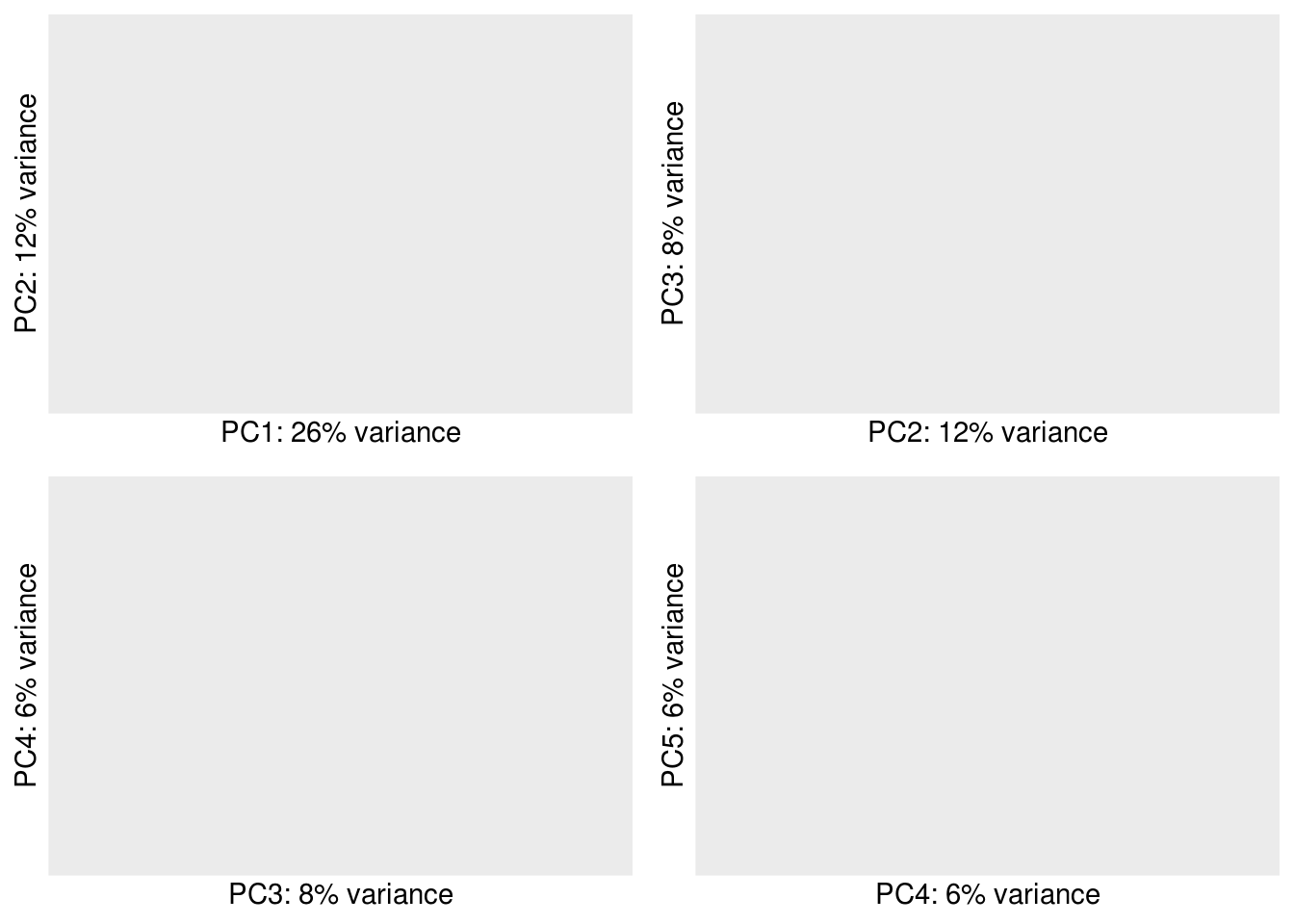
3.1.3 Merging Replicate and Data Prep for Downstream Anakysis
dds_c <- collapseReplicates(dds, dds$name)
ntd <- normTransform(dds_c)
vsd <- vst(dds_c, blind=FALSE) msd.ntd <- meanSdPlot(assay(ntd), plot = F)
msd.vsd <- meanSdPlot(assay(vsd), plot = F)
# msd.rld <- meanSdPlot(assay(rld), plot = F)
ymax <- max(c(max(msd.ntd$gg$data$py),
max(msd.vsd$gg$data$py)))#,
#max(msd.rld$gg$data$py)))
p <- list()
p[[1]] <- msd.ntd$gg + ggtitle('log2(n + 1)') + ylim(c(0,ymax))
p[[2]] <- msd.vsd$gg + ggtitle('Variance stabilizing transformation') + ylim(c(0,ymax))
# p[[3]] <- msd.rld$gg + ggtitle('Regularized log transformation') + ylim(c(0,ymax))
library(cowplot)
plot_grid(plotlist = p, align = 'hv', ncol = 2)#3)## Warning: Removed 8 rows containing missing values (geom_hex).## Warning: Removed 11 rows containing missing values (geom_hex).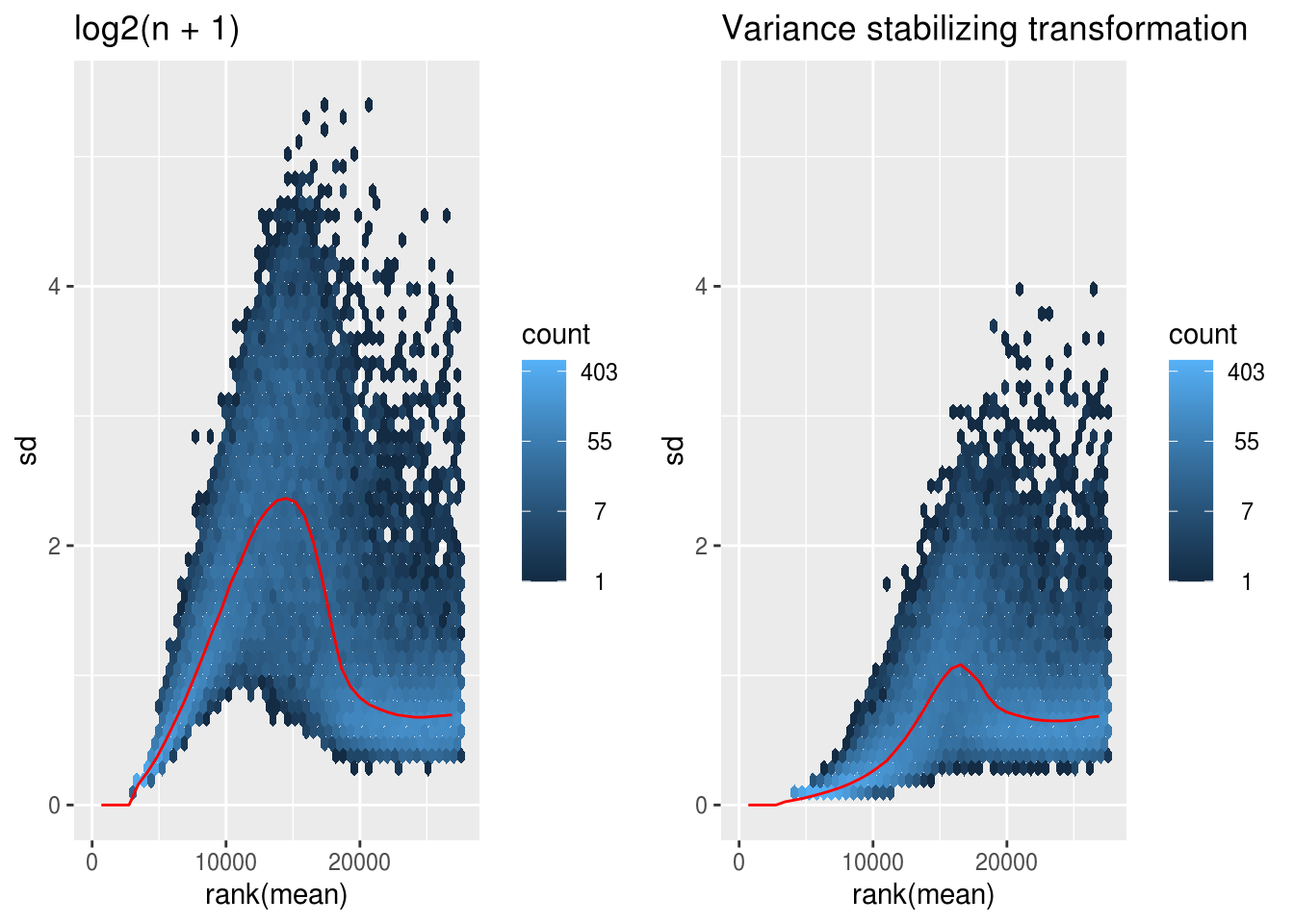
3.1.3.1 Correlation plot for Merged Data
M <- counts(dds_c)
colnames(M) <- sapply(colnames(M), function(x){ y <- str_replace_all(x," [0-9]$","")})
corrplot(cor(M), # Correlation matrix
method = "square", # Correlation plot method
type = "full", # Correlation plot style (also "upper" and "lower")
diag = TRUE, # If TRUE (default), adds the diagonal
tl.col = "black", # Labels color
bg = "white", # Background color
order = "hclust",
hclust.method= "ward.D2",
tl.cex = 0.8,
col = NULL) # Color palette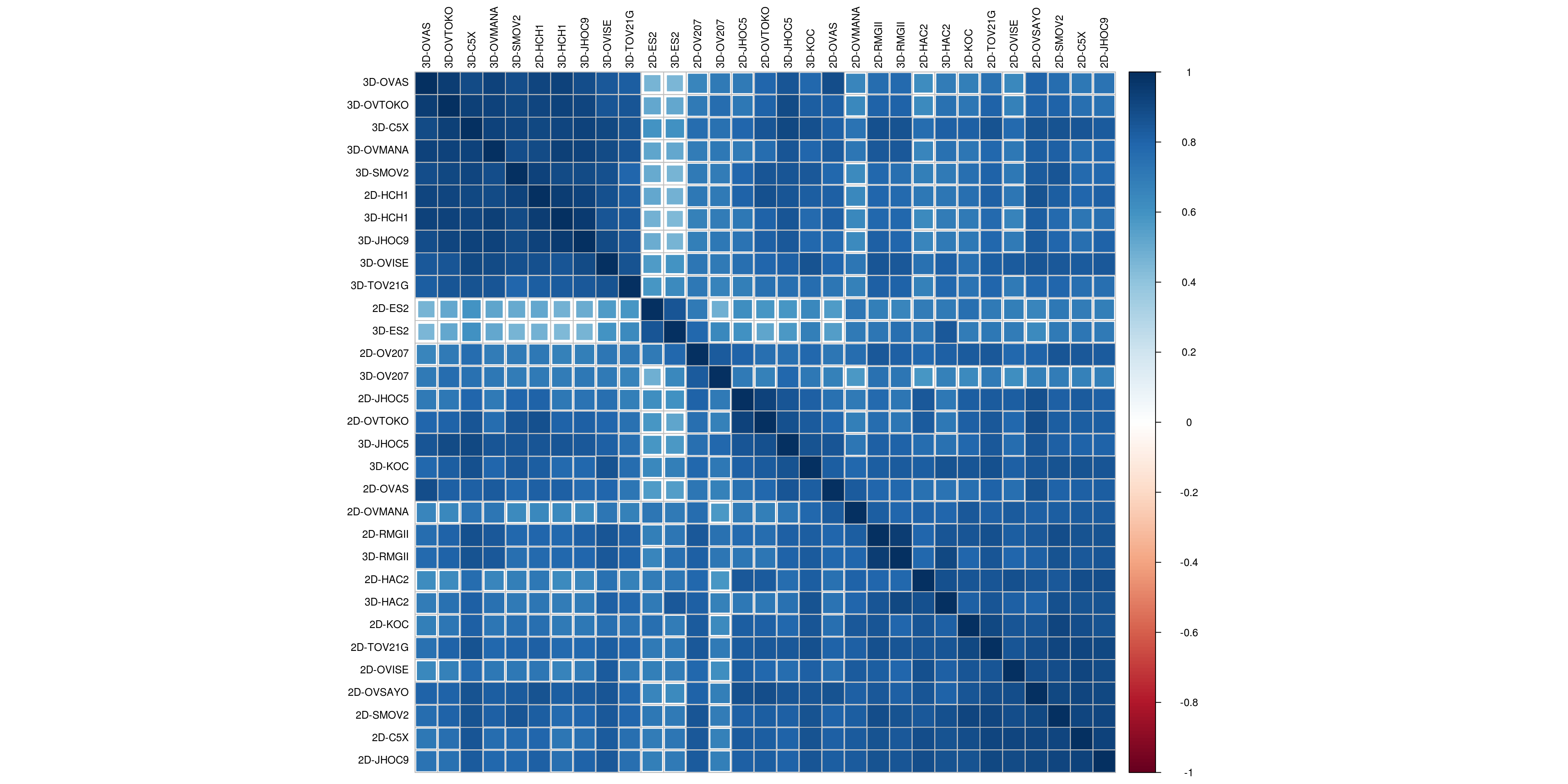
3.1.3.2 Sample to Sample Distance for Merged Data
sampleDists <- dist(t(assay(vsd)))
sampleDistMatrix <- sampleDists %>% as.matrix %>% as.data.frame
rownames(sampleDistMatrix) <- vsd$name
colnames(sampleDistMatrix) <- vsd$name
colors <- heat.map.color <- viridis(n = 256, alpha = 1, begin = 0, end = 1, option = "mako")
pheatmap(sampleDistMatrix,
clustering_distance_rows=sampleDists,
clustering_distance_cols=sampleDists,
clustering_method = 'ward.D2',
col=colors, fontsize = 8, fontsize_row = 4)3.1.3.3 PCA for Merged Data
show.labels <- TRUE # change to TRUE if you want to show the sample IDs
pcaData <- plotPCA.DESeqTransform(vsd, intgroup='type', returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
# adding cell line name into pcaData dataframe
pcaData <- merge(pcaData, unique(dds_c@colData[c("name","cell.line")]) , by="name") %>% as.data.frame
if(show.labels){
#label <- pcaData$cell.name
label <- pcaData$name
}else{
label <- NA
}
PC.list <-c(1:4)
g.plot <- lapply( PC.list , function(i){
x=paste("PC",i,sep="")
y=paste("PC",i+1,sep="")
return( ggplot(pcaData[c(x,y,"cell.line","type")], aes( .data[[x]], .data[[y]], color= cell.line ,shape=type ,label = label)) +
geom_point(size=2) +
xlab(paste0(x,": ",percentVar[i],"% variance")) +
ylab(paste0(y,": ",percentVar[i+1],"% variance")) +
geom_text_repel(show.legend = F,size=2)+
guides(colour="none"))+
theme_minimal()
})
plot_grid(plotlist = g.plot, align = "h")## Warning: ggrepel: 4 unlabeled data points (too many overlaps). Consider increasing max.overlaps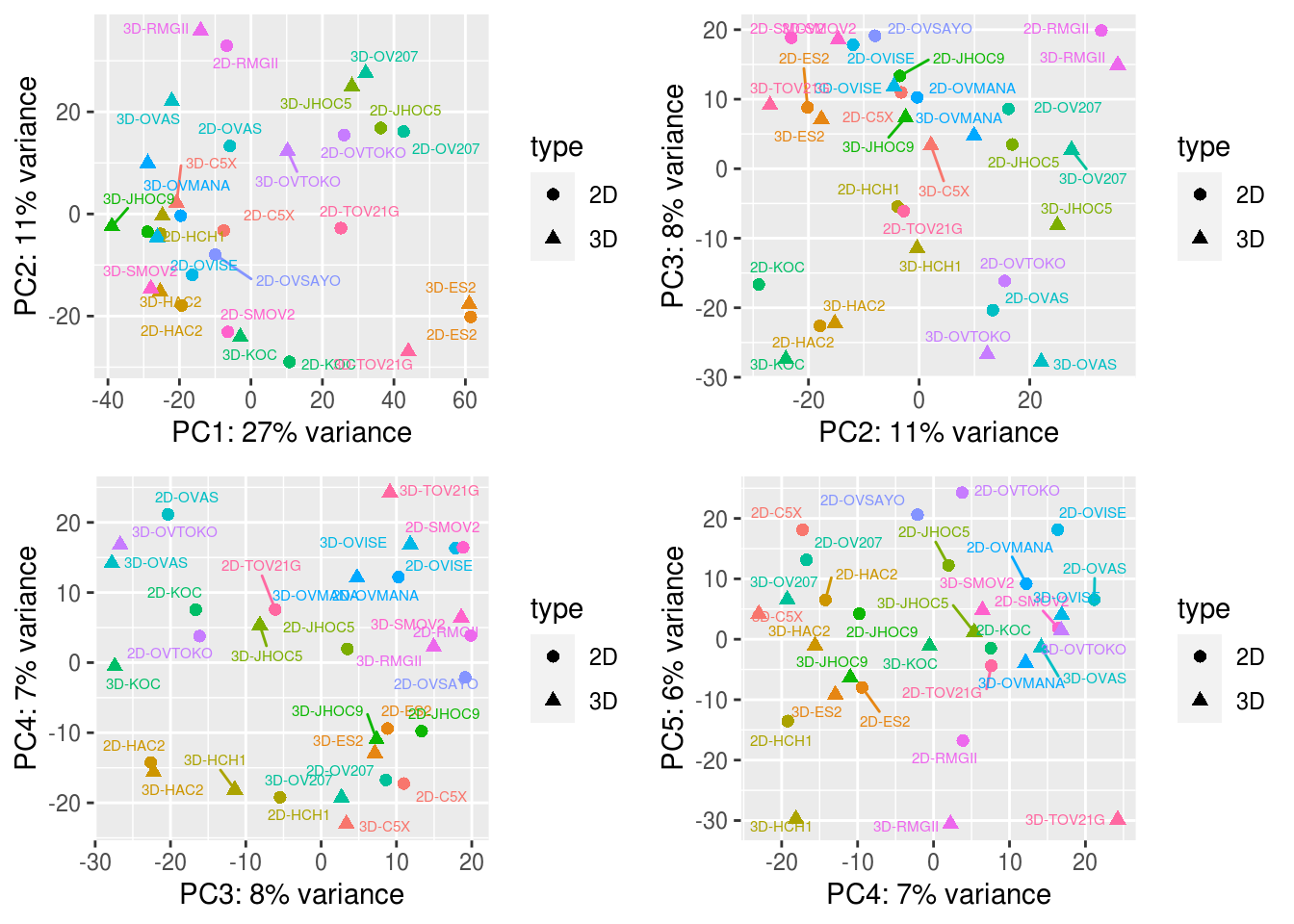
3.2 2D Consensus clustering
3.2.1 Data Preparation
df <- as.data.frame(colData(dds_c)[,c("cell.line","type")]) %>% filter(type =="2D")%>%mutate(rowname= rownames(.))
df <- merge(df, CCOC_mutation_table[,c(1,3:12)] %>% dplyr::select(where(~ any(. != 0))), by.x="cell.line", by.y="cell_line", all.x=TRUE)
df <- merge(df, CCOC.Gly, by.x="cell.line", by.y="cell_line", all.x=TRUE ) %>% magrittr::set_rownames(.$rowname) %>% dplyr::select(-rowname)%>% dplyr::mutate( across(where(is.numeric), ~coalesce(., 0)))
ann_colors <- list(type= c(`3D`= "#1B9E77",`2D` = "#D95F02"))
vsd.gene.2D <- assay(vsd) %>% as.data.frame %>% dplyr::select(., starts_with("2D-")) %>% as.matrix
heat.map.color <- viridis(n = 256, alpha = 1, begin = 0, end = 1, option = "viridis")
#getting meta dataframe for 2D data
df <- get.2D.pheatmap.df()col.list <- c(50, 500, 1000, 5000)
dds_c.mtx.select <- counts(dds_c,normalized=TRUE) %>% as.data.frame %>% dplyr::select(., starts_with(c("2D-","CCOC_"))) %>% as.matrix
pmap.df.list <- lapply(col.list, function(i){
str_glue("#### Top **{i}** SD Gene ")
select <- order(rowSds(dds_c.mtx.select),decreasing=TRUE)[1:i]
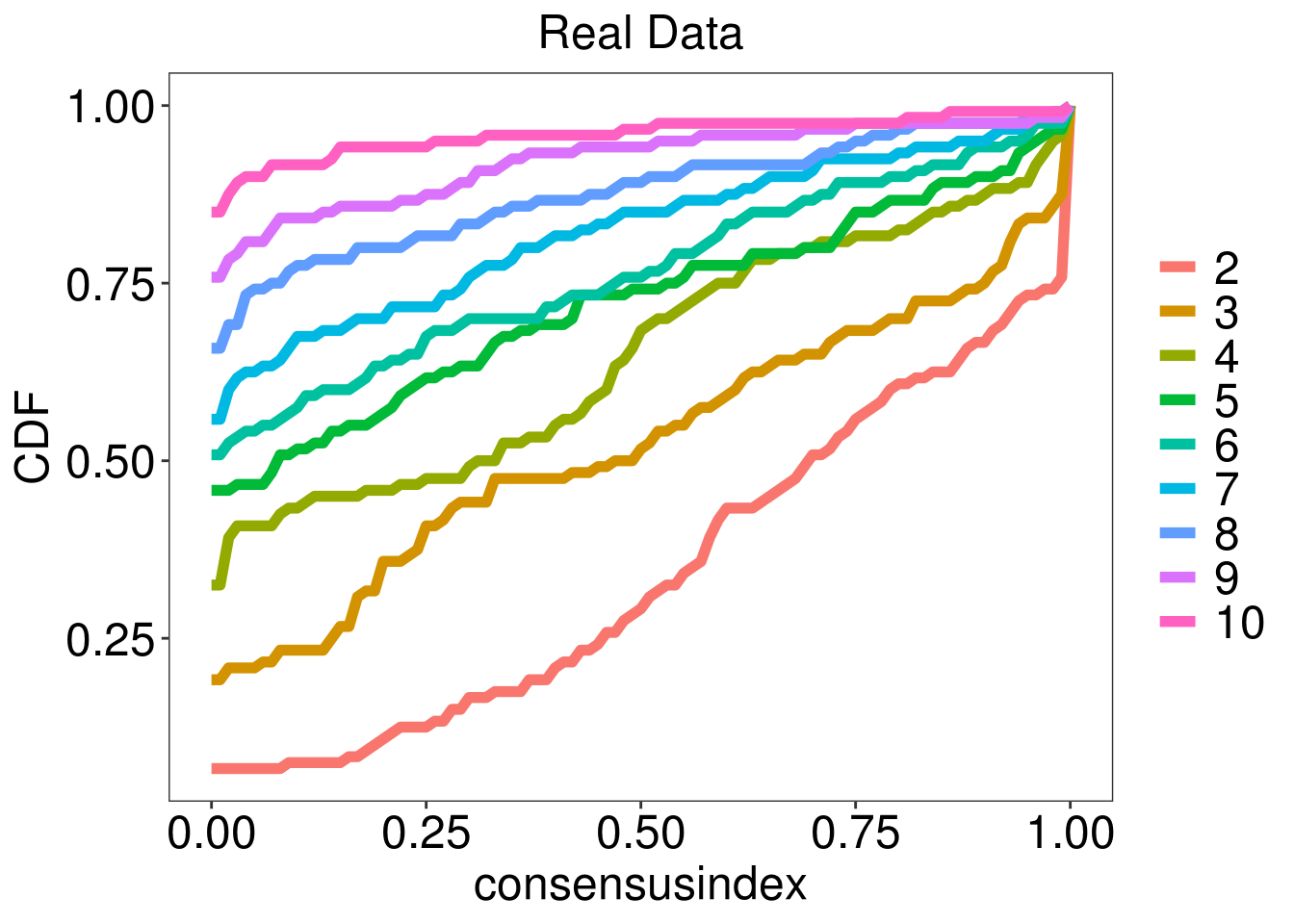
test <- M3C(data.frame(vsd.gene.2D[select,]))
k <- test$scores %>% mutate_all(as.numeric) %>% slice(which.max(RCSI)) %>% dplyr::select(K) %>% as.numeric
cluster <- test$realdataresults[[k]]$ordered_annotation
c.name <- factor( rownames(cluster) , labels=colnames(dds_c.mtx.select),levels=colnames(data.frame(vsd.gene.2D[select,])))
rownames(cluster) <- c.name
df <- merge(df,cluster, by.y="row.names" , by.x="row.names") %>% arrange(consensuscluster)
rownames(df)<- df$Row.names
heat.map.ann.color <- viridis(n = k, alpha = 1, begin = 0, end = 1, option = "viridis")
heat.map.ann.color.map <- unique(df$consensuscluster)
heat.map.ann.color<- setNames(heat.map.ann.color.map,heat.map.ann.color.map)
annotation_colors = list(consensuscluster=heat.map.ann.color)
pmap <- pheatmap(vsd.gene.2D[select,], cluster_rows=TRUE, treeheight_row = 0, show_rownames=FALSE,
cluster_cols=TRUE,color=heat.map.color , annotation_col=df, annotation_colors =annotation_colors ,fontsize = 8, annotation_legend = FALSE ,cutree_cols=k,clustering_method ="ward.D2")[[4]]
return(structure(pmap,cluster=cluster))
})
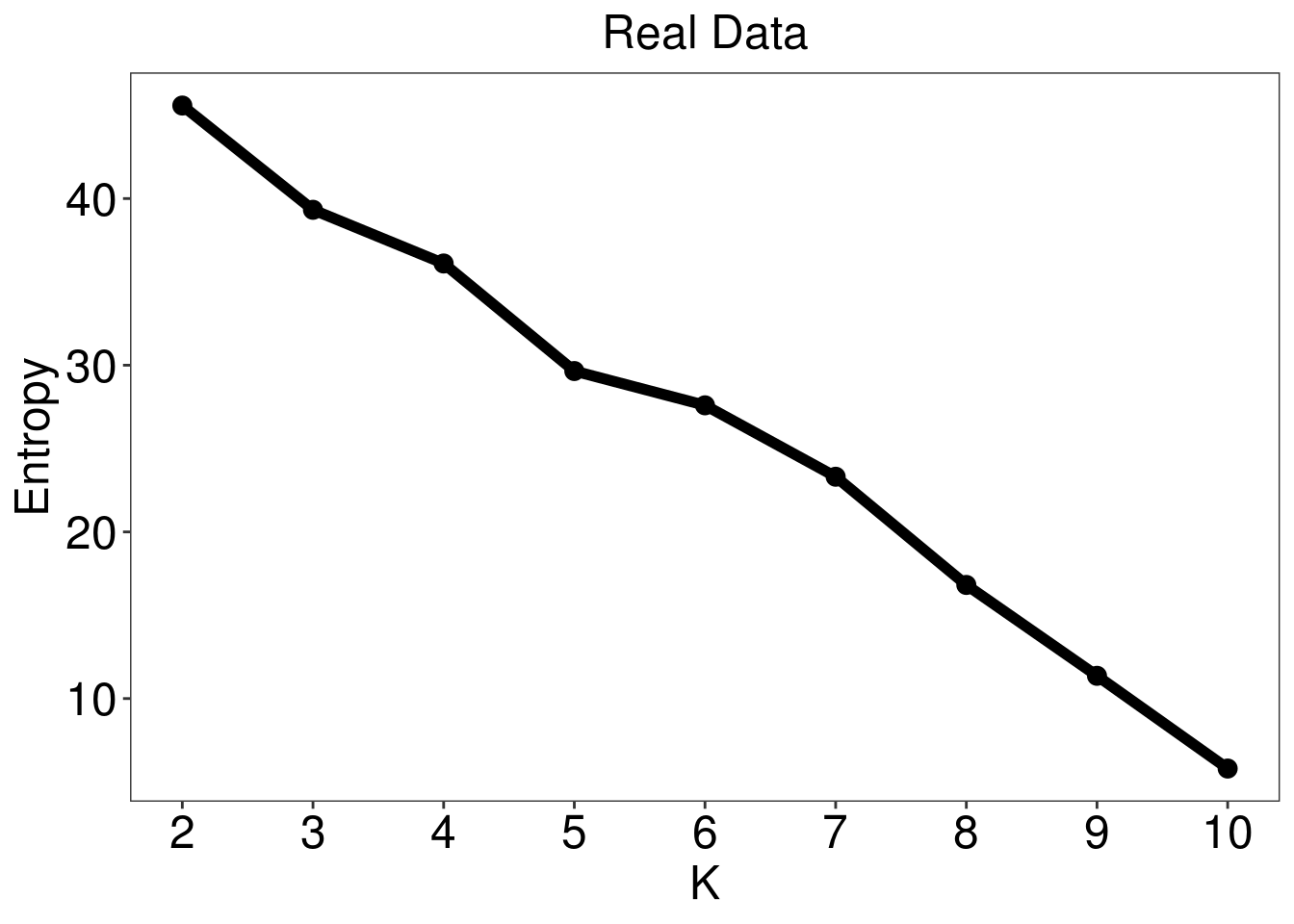
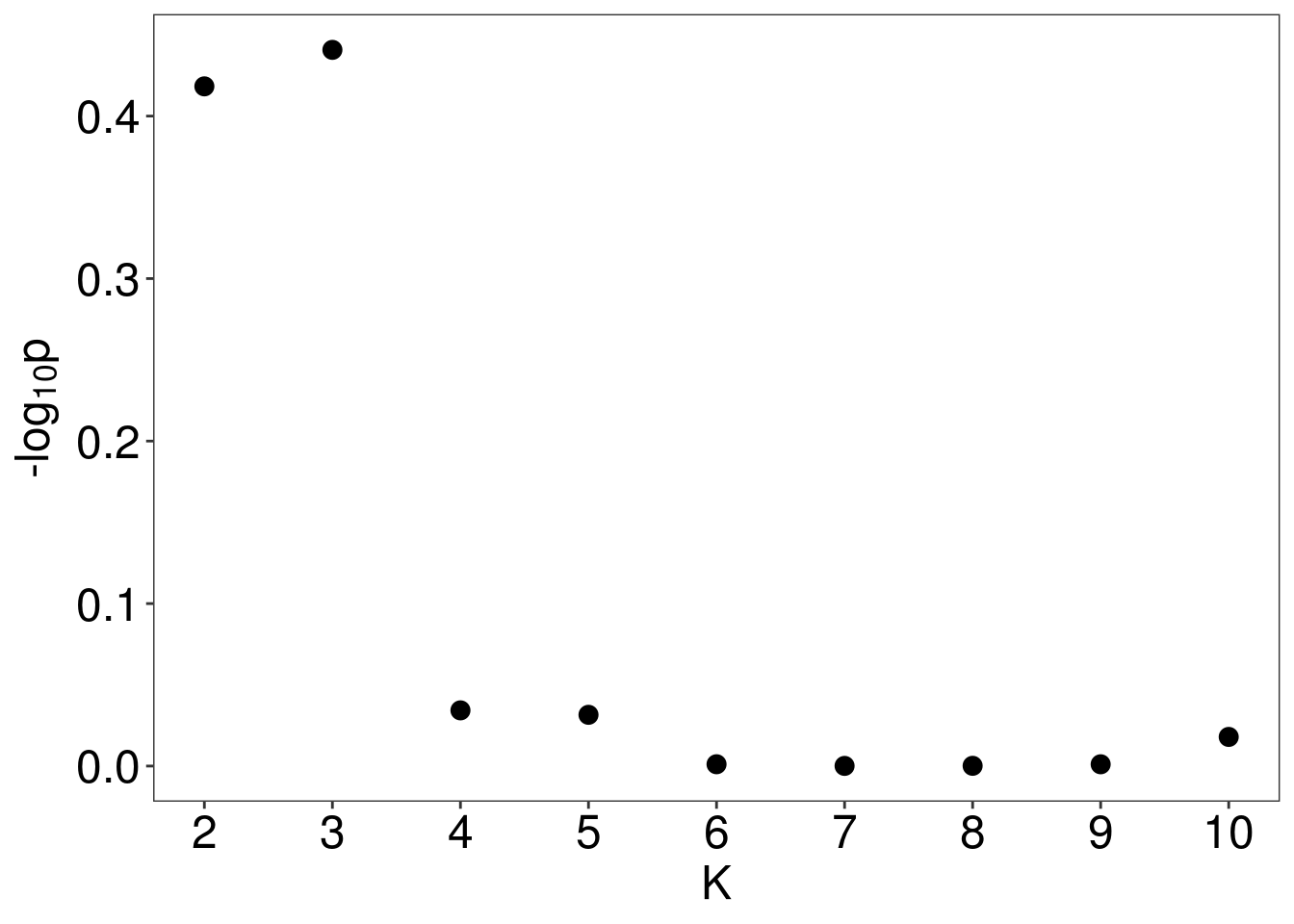
3.2.2 Expression of PI3CKA, ARID1A, HNF1B for consensus clustering of Top 5000 variable gene
cluster.assignment <- attr(pmap.df.list[[4]],"cluster")
cluster.expression.volin.df <- vsd.gene.2D %>% as.data.frame %>% dplyr::filter(str_detect(row.names(vsd.gene.2D),"PIK3CA|PIK3R1|ARID1A|HNF1B")) %>% rownames_to_column() %>% mutate(rowname=str_replace(rowname,"^.+_","")) %>% reshape2::melt() %>% merge(.,cluster.assignment,by.x="variable",by.y=0)## Using rowname as id variablesxlabs <- paste(unique(cluster.expression.volin.df$consensuscluster),"\n(N=",table(cluster.expression.volin.df$consensuscluster)/length(unique(cluster.expression.volin.df$rowname)),")",sep="")
ggplot(cluster.expression.volin.df, aes(x=consensuscluster, y=value)) +geom_boxplot(aes(fill = rowname),width=0.5,position = position_dodge(0.8))+ geom_dotplot(aes(fill = rowname),binaxis = "y", stackdir='center', dotsize = 0.8,position = position_dodge(0.8))+ scale_x_discrete(labels=xlabs)+labs(y="Gene Epression Level (vsd)") + theme_classic()## Bin width defaults to 1/30 of the range of the data. Pick better value with `binwidth`.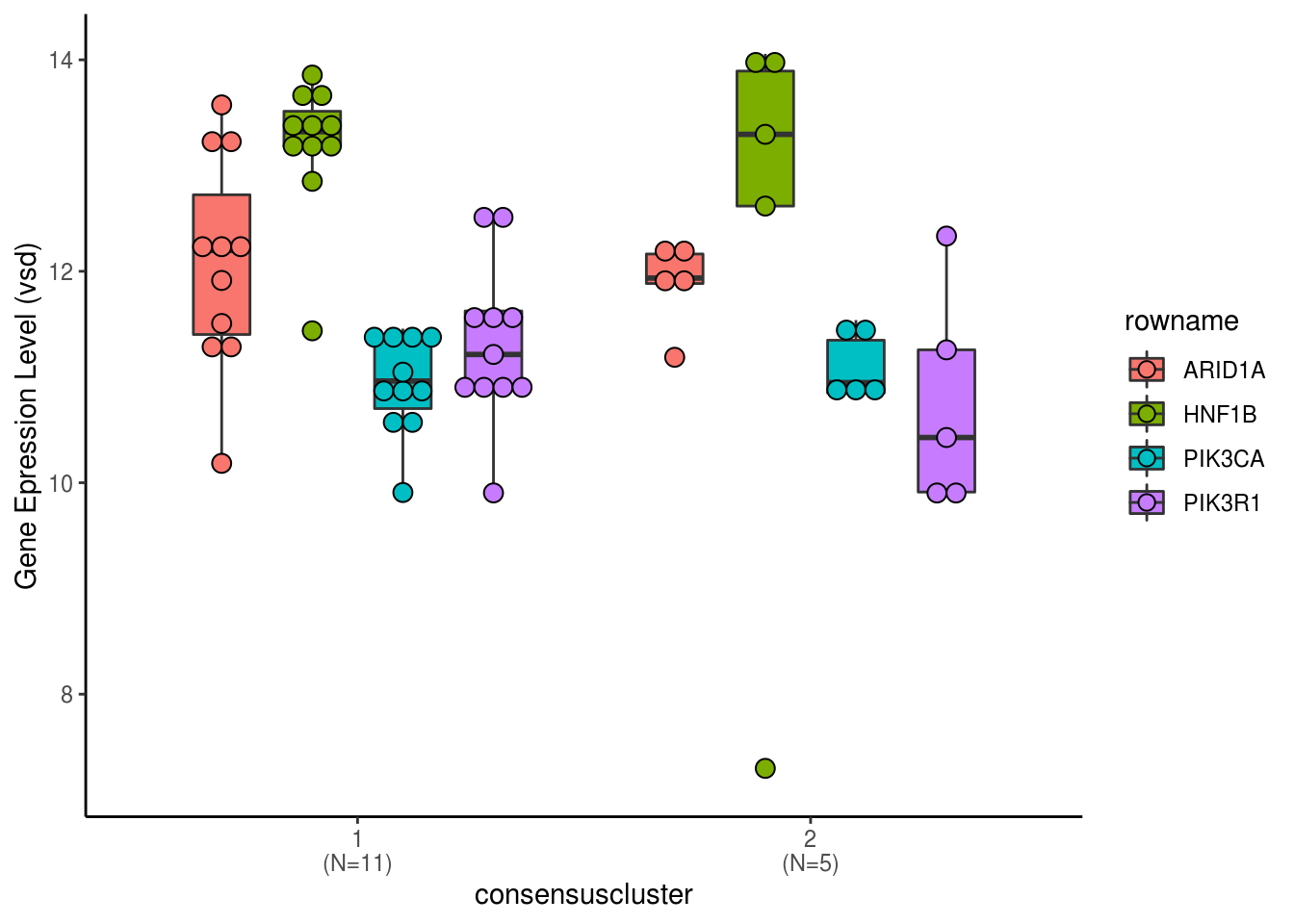
3.2.3 Global Heatmap for Consensus Clustering of Top 50, 500, 1000, 5000 variable genes
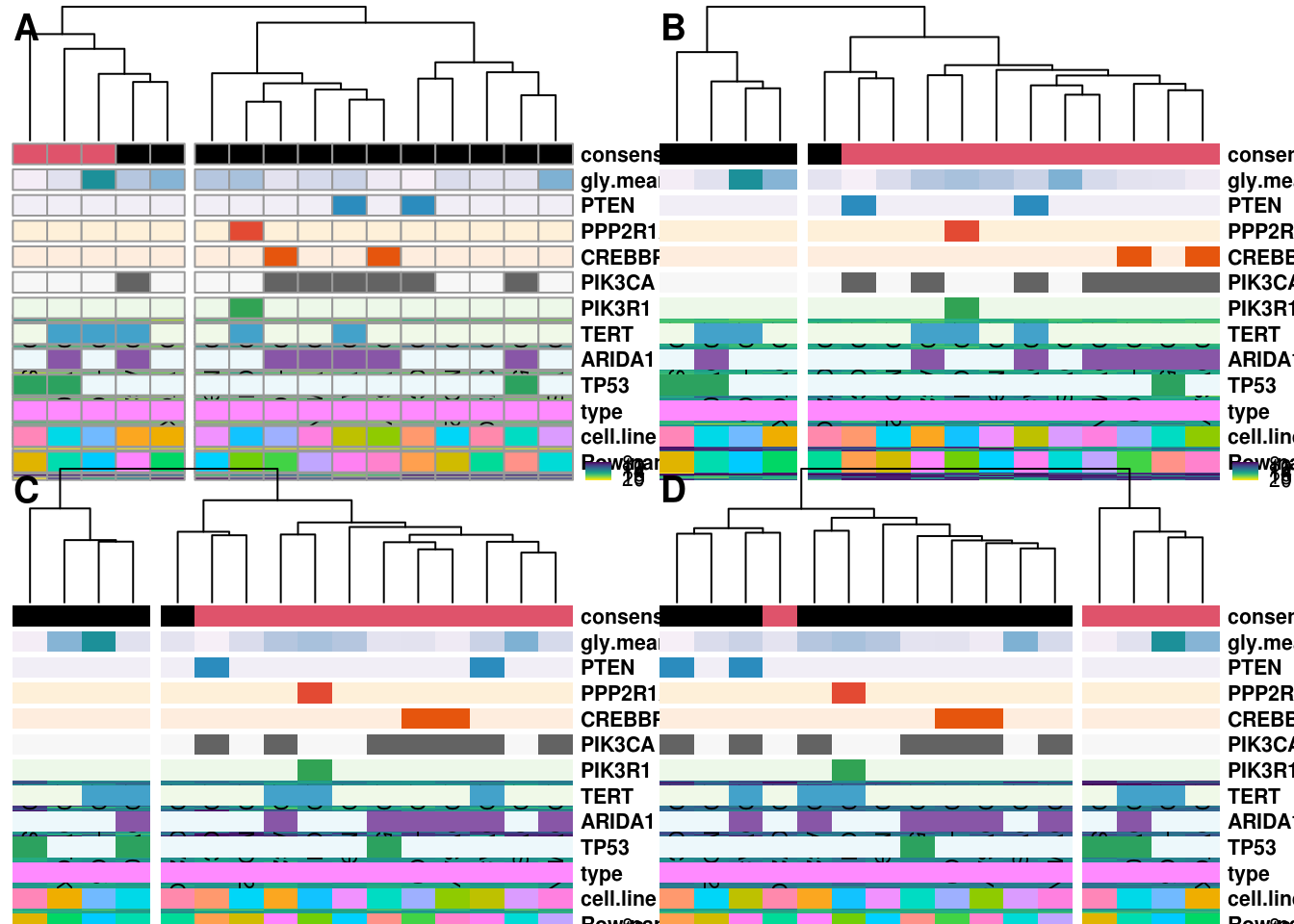
3.2.4 2D Consensus clustering DGE
dds.2D <- dds[,colData(dds)$type == "2D"]
colData(dds.2D) <- merge(colData(dds.2D),attributes(pmap.df.list[[4]])$cluster, by.x="name", by.y=0)
dds_c.2D.df <- merge(colData(dds_c),attributes(pmap.df.list[[4]])$cluster, by.x="name", by.y=0) %>% as.data.frame
dds_c.2D.df <- merge(dds_c.2D.df,CCOC_mutation_table[,c(1,3:12)],by.x="cell.line", by.y="cell_line",all.x=TRUE)
dds_c.2D.df <- dds_c.2D.df %>% mutate(PIK3=PIK3R1+PIK3CA+PTEN) %>% dplyr::select(-c(PIK3R1,PIK3CA,PTEN))
dds_c.2D.df <- merge(dds_c.2D.df,CCOC.Gly,by.x="cell.line", by.y="cell_line",all.x=TRUE)%>% column_to_rownames("name")
design(dds.2D) <- ~ cell.batch + consensuscluster
dds.2D <- estimateSizeFactors(dds.2D)
dds.2D <- DESeq(dds.2D)alpha <- 0.01
# get FC and pvalues (equivalent methods)
res.2D <- results(dds.2D, name="consensuscluster_2_vs_1", alpha = alpha)
resLFC.2D <- lfcShrink(dds.2D, coef = 3, type="apeglm")
resNorm.2D <- lfcShrink(dds.2D, coef = 3, type="normal")
resAsh.2D <- lfcShrink(dds.2D, coef = 3, type="ashr")
par(mfrow=c(1,4), mar=c(4,4,2,1))
yMax <- max(abs(c(res.2D$log2FoldChange, resLFC.2D$log2FoldChange, resNorm.2D$log2FoldChange, resAsh.2D$log2FoldChange)), na.rm = TRUE)
xMax <- max(c(plotMA(object = res.2D, returnData = T)[,1], plotMA(object = resLFC.2D, returnData = T)[,1], plotMA(object = resNorm.2D, returnData = T)[,1], plotMA(object = resAsh.2D, returnData = T)[,1]))
xlim <- c(1,xMax); ylim <- c(-yMax, yMax)
plotMA(res.2D, xlim=xlim, ylim=ylim, main="none", alpha = alpha) # default alpha = 0.10
plotMA(resLFC.2D, xlim=xlim, ylim=ylim, main="apeglm", alpha = alpha) # default alpha = 0.10
plotMA(resNorm.2D, xlim=xlim, ylim=ylim, main="normal", alpha = alpha) # default alpha = 0.10
plotMA(resAsh.2D, xlim=xlim, ylim=ylim, main="ashr", alpha = alpha) # default alpha = 0.10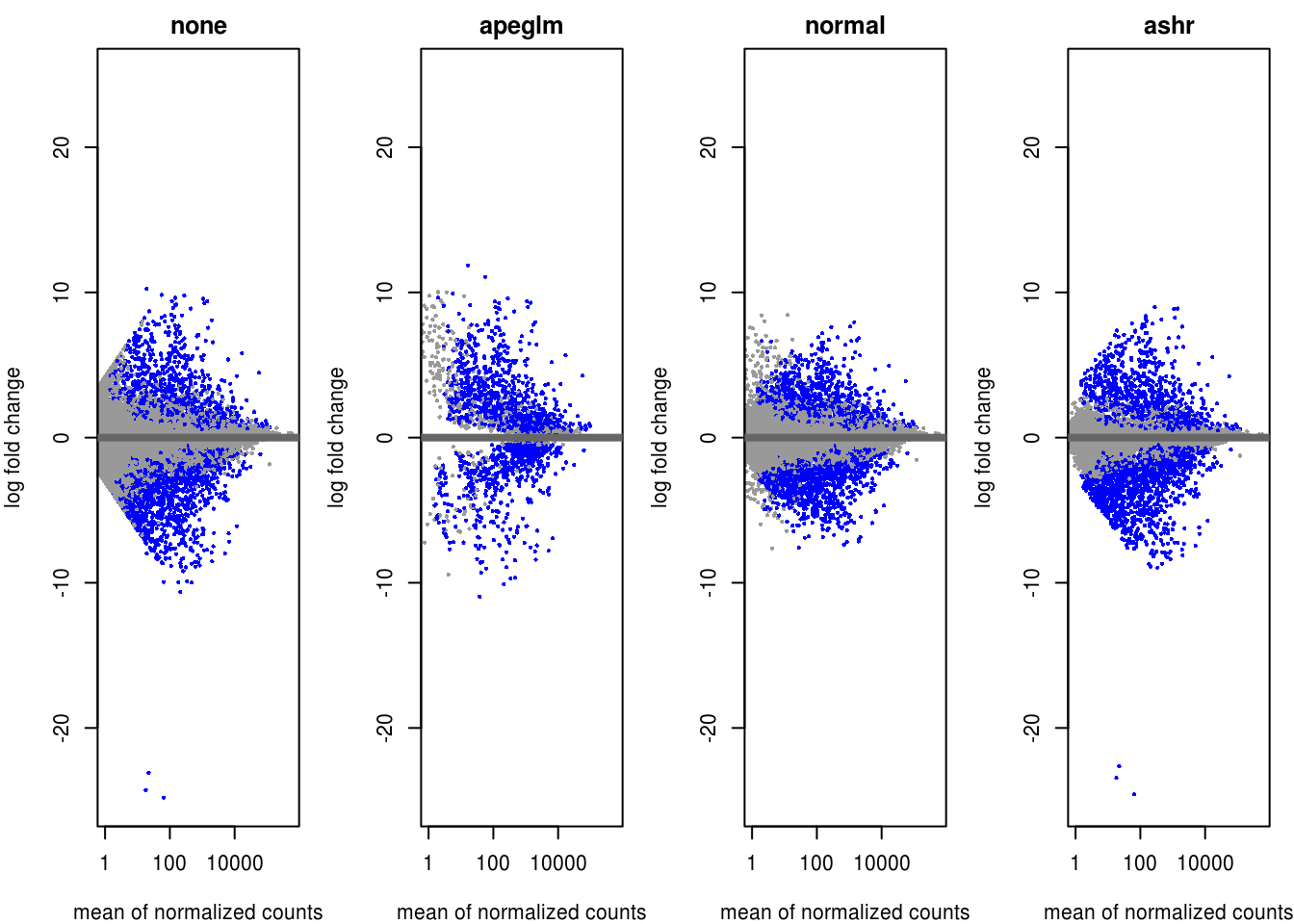
lsf.volcano <- lapply( list(res.2D,resLFC.2D,resNorm.2D,resAsh.2D), function(x){
ggplot(data=data.frame(x) %>% na.omit %>% filter(abs(log2FoldChange) < 15), aes(x=log2FoldChange, y=-log10(padj))) +
geom_point() +
theme_minimal() +
geom_vline(xintercept=c(-0.6, 0.6), col="red") +
geom_hline(yintercept=-log10(alpha), col="red") +
xlim(-15,15) +
ylim(-5,60)
})
#do.call("grid.arrange", c(lsf.volcano, ncol=2, nrow=2))
do.call("plot_grid",c(lsf.volcano, labels ="auto"))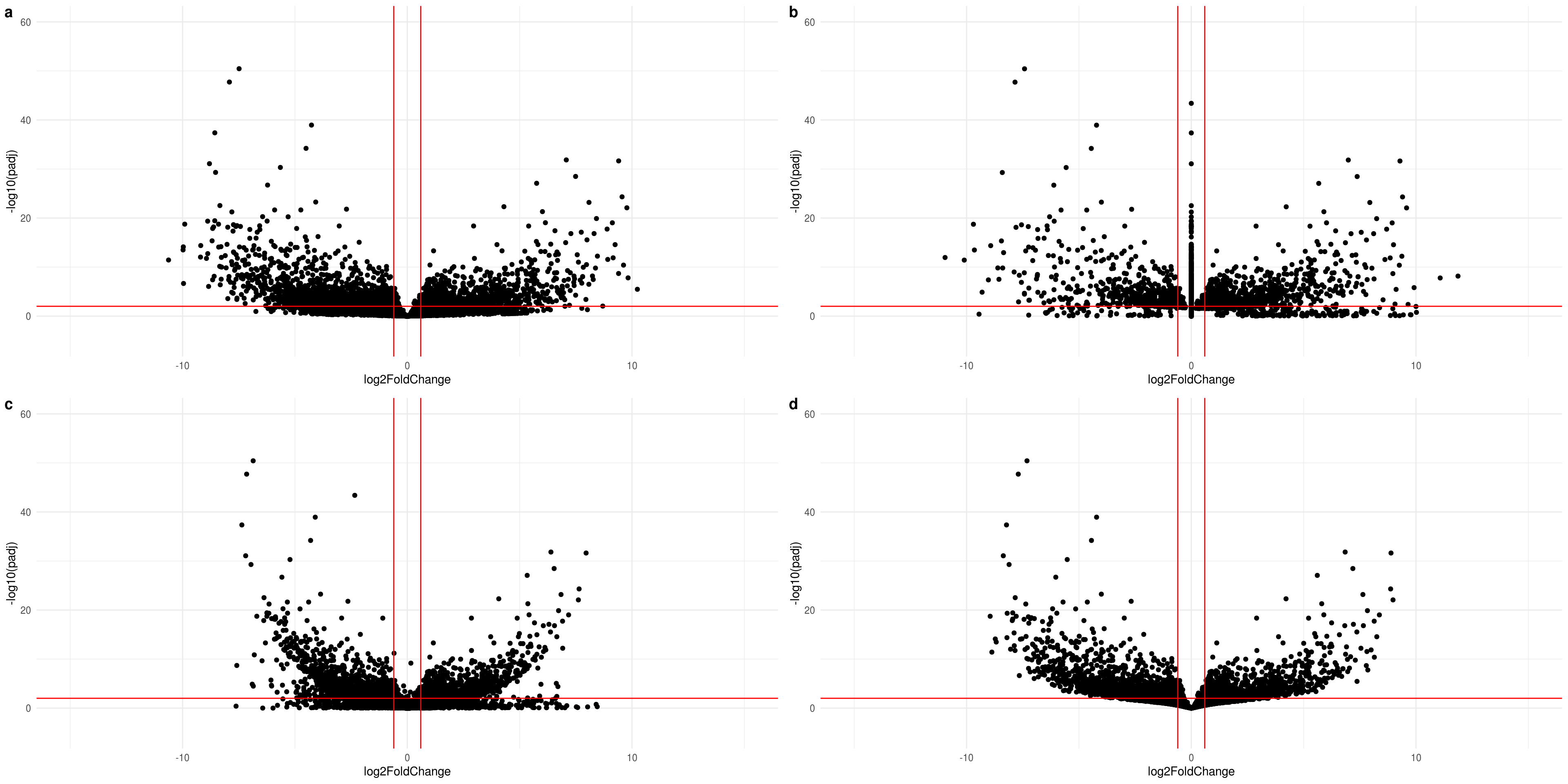
resAsh.2D <- resAsh.2D[resAsh.2D$padj < alpha & !(is.na(resAsh.2D$padj)),]
resOrdered <- resAsh.2D %>% as.data.frame %>% rownames_to_column %>% arrange(padj, desc(abs(resAsh.2D$log2FoldChange)))
resOrdered.up <- resOrdered %>% filter(resAsh.2D$log2FoldChange > 0)
resOrdered.down <- resOrdered %>% filter(resAsh.2D$log2FoldChange < 0)
#p.value.max <- max(resOrdered$padj[1:50])
#FC.min <- max(resOrdered$log2FoldChange[1:50])
#p.value.max <- max(resOrdered.down$padj[1:50])
#select <- which( (abs(df.resAsh$log2FoldChange < 0) & df.resAsh$padj <= p.value.max & !(is.na(df.resAsh$padj))) )
select <- which(rownames(vsd.gene.2D) %in% resOrdered$rowname & str_replace(rownames(vsd.gene.2D),"\\..*$","") %in% protein.gene$ensembl_gene_id )
pheatmap(vsd.gene.2D[select,], cluster_rows=TRUE, show_rownames=FALSE,color=heat.map.color,
cluster_cols=TRUE, annotation_col=dds_c.2D.df["consensuscluster"],fontsize = 7,clustering_method ="ward.D2")3.2.4.1 Top 50 Genes Heatmap
select <- select[1:50]
heatmap.df<- dds_c.2D.df %>% dplyr::select(-c(type,sizeFactor,type,cell.line,cell.batch))
rownames(vsd.gene.2D) <- str_replace(rownames(vsd.gene.2D),"^.+_","")
pheatmap(vsd.gene.2D[select,order(heatmap.df$consensuscluster)], cluster_rows=TRUE,color=heat.map.color,
cluster_cols=FALSE, treeheight_row=0, annotation_col=heatmap.df,fontsize = 7,clustering_method ="ward.D2")3.3 Differential gene expression
3.3.1 Cell meduim - 3D vs 2D
## using pre-existing size factors## estimating dispersions## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testing## 5 rows did not converge in beta, labelled in mcols(object)$betaConv. Use larger maxit argument with nbinomWaldTest3.3.1.1 Log fold change shrinkage
alpha <- 0.01
# get FC and pvalues (equivalent methods)
res <- results(dds, name="type_3D_vs_2D", alpha = alpha)
resLFC <- lfcShrink(dds, coef = 2, type="apeglm")
resNorm <- lfcShrink(dds, coef = 2, type="normal")
resAsh <- lfcShrink(dds, coef = 2, type="ashr")
par(mfrow=c(1,4), mar=c(4,4,2,1))
yMax <- max(abs(c(res$log2FoldChange, resLFC$log2FoldChange, resNorm$log2FoldChange, resAsh$log2FoldChange)), na.rm = TRUE)
xMax <- max(c(plotMA(object = res, returnData = T)[,1], plotMA(object = resLFC, returnData = T)[,1], plotMA(object = resNorm, returnData = T)[,1], plotMA(object = resAsh, returnData = T)[,1]))
xlim <- c(1,xMax); ylim <- c(-yMax, yMax)
plotMA(res, xlim=xlim, ylim=ylim, main="none", alpha = alpha) # default alpha = 0.10
plotMA(resLFC, xlim=xlim, ylim=ylim, main="apeglm", alpha = alpha) # default alpha = 0.10
plotMA(resNorm, xlim=xlim, ylim=ylim, main="normal", alpha = alpha) # default alpha = 0.10
plotMA(resAsh, xlim=xlim, ylim=ylim, main="ashr", alpha = alpha) # default alpha = 0.10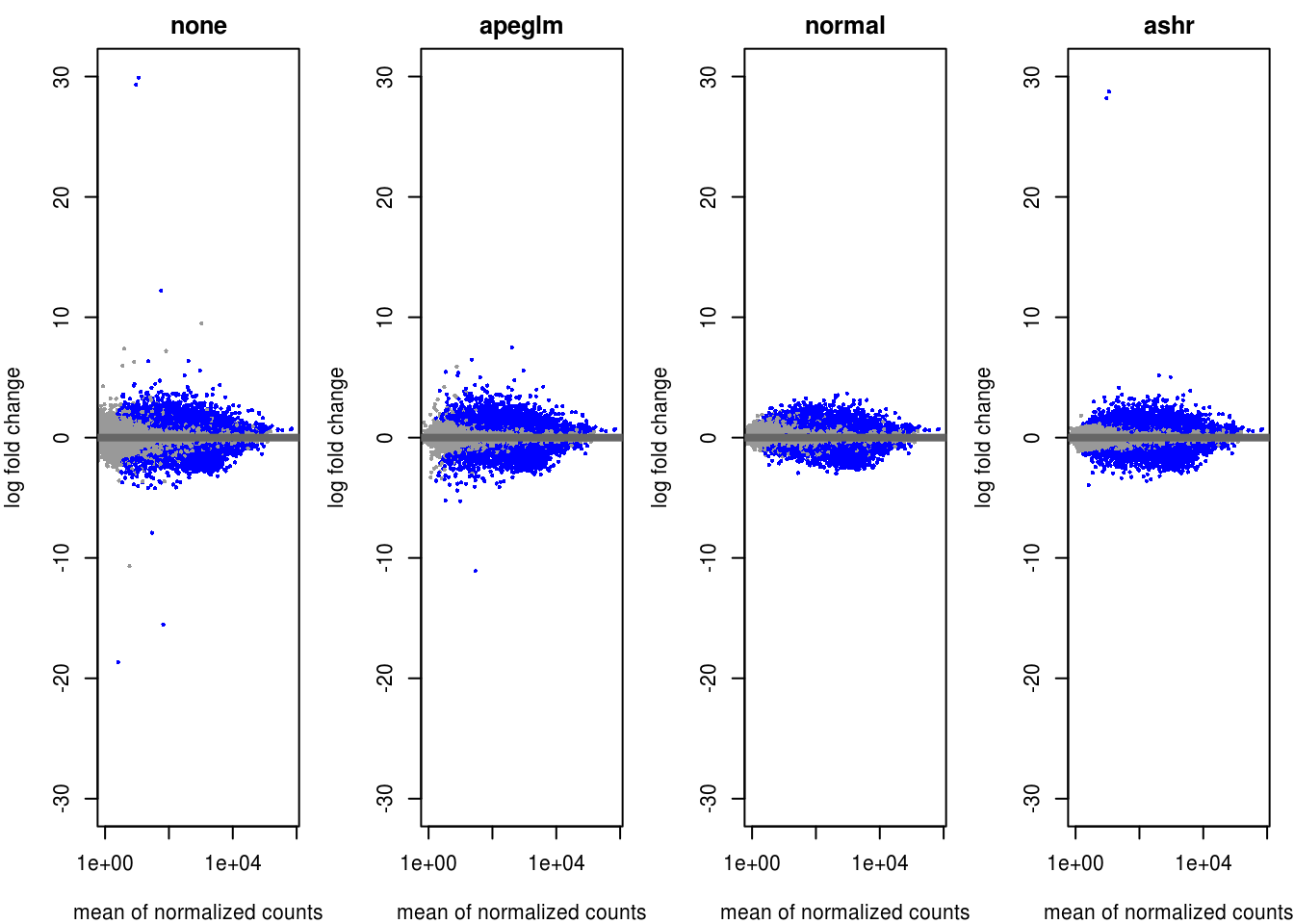
lsf.volcano <- lapply( list(res,resLFC,resNorm,resAsh), function(x){
ggplot(data=data.frame(x) %>% na.omit %>% filter(abs(log2FoldChange) < 15), aes(x=log2FoldChange, y=-log10(padj))) +
geom_point() +
theme_minimal() +
geom_vline(xintercept=c(-0.6, 0.6), col="red") +
geom_hline(yintercept=-log10(alpha), col="red") +
xlim(-15,15) +
ylim(-5,60)
})
#do.call("grid.arrange", c(lsf.volcano, ncol=2, nrow=2))
do.call("plot_grid",c(lsf.volcano, labels ="auto"))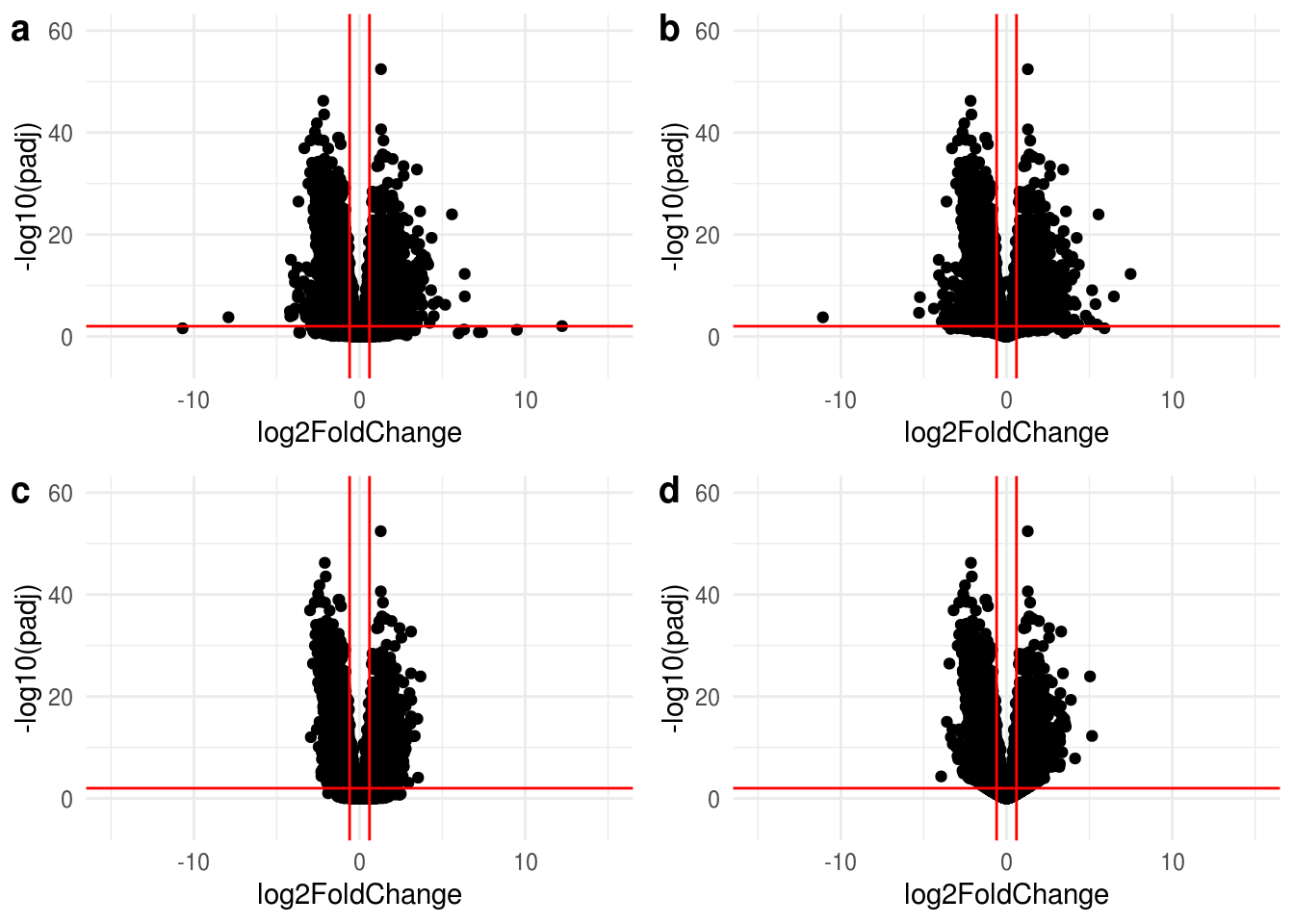
3.3.2 Cell lines Comparison
## Note: levels of factors in the design contain characters other than
## letters, numbers, '_' and '.'. It is recommended (but not required) to use
## only letters, numbers, and delimiters '_' or '.', as these are safe characters
## for column names in R. [This is a message, not a warning or an error]## using pre-existing size factors## estimating dispersions## found already estimated dispersions, replacing these## gene-wise dispersion estimates## mean-dispersion relationship## Note: levels of factors in the design contain characters other than
## letters, numbers, '_' and '.'. It is recommended (but not required) to use
## only letters, numbers, and delimiters '_' or '.', as these are safe characters
## for column names in R. [This is a message, not a warning or an error]## final dispersion estimates## Note: levels of factors in the design contain characters other than
## letters, numbers, '_' and '.'. It is recommended (but not required) to use
## only letters, numbers, and delimiters '_' or '.', as these are safe characters
## for column names in R. [This is a message, not a warning or an error]## fitting model and testing## 5 rows did not converge in beta, labelled in mcols(object)$betaConv. Use larger maxit argument with nbinomWaldTestalpha <- 0.01
threshold <- 1
contrast.group <- ddsgrp@colData %>% as.data.frame %>% dplyr::select(-c(cell.batch,sizeFactor)) %>% mutate_at("name",as.factor) %>% unique() %>% group_by(cell.line) %>% group_split(name)
#res.list <- lapply(contrast.group[-c(12)], function(x){ x<- unlist(as.character(x$name)) ; results(ddsgrp,contrast = c("name",x),alpha = alpha)})
res.ashr.list <- lapply(contrast.group[-c(12)], function(x){ x<- unlist(as.character(x$name)) ; lfcShrink(ddsgrp,contrast = c("name",x), type="ashr")})yMax <- quantile(sapply(res.ashr.list,function(x){max(abs(x$log2FoldChange),na.rm= TRUE)}),0.75)
yMax <- 10
yMax.padj <- quantile(sapply(res.ashr.list,function(x){max(-log10(x$padj[x$padj >0]),na.rm= TRUE)}),0.85)
xMax <- max(sapply(res.ashr.list,function(x){plotMA(object = x, returnData = T)[,1]}))
xlim <- c(1,xMax); ylim <- c(-yMax, yMax)
volcano.plots <- lapply(res.ashr.list, function(x){plot.volcano(x,ylim,c(-yMax.padj*0.2,yMax.padj),5)})
do.call("plot_grid",c(volcano.plots, labels ="auto", nrow=6))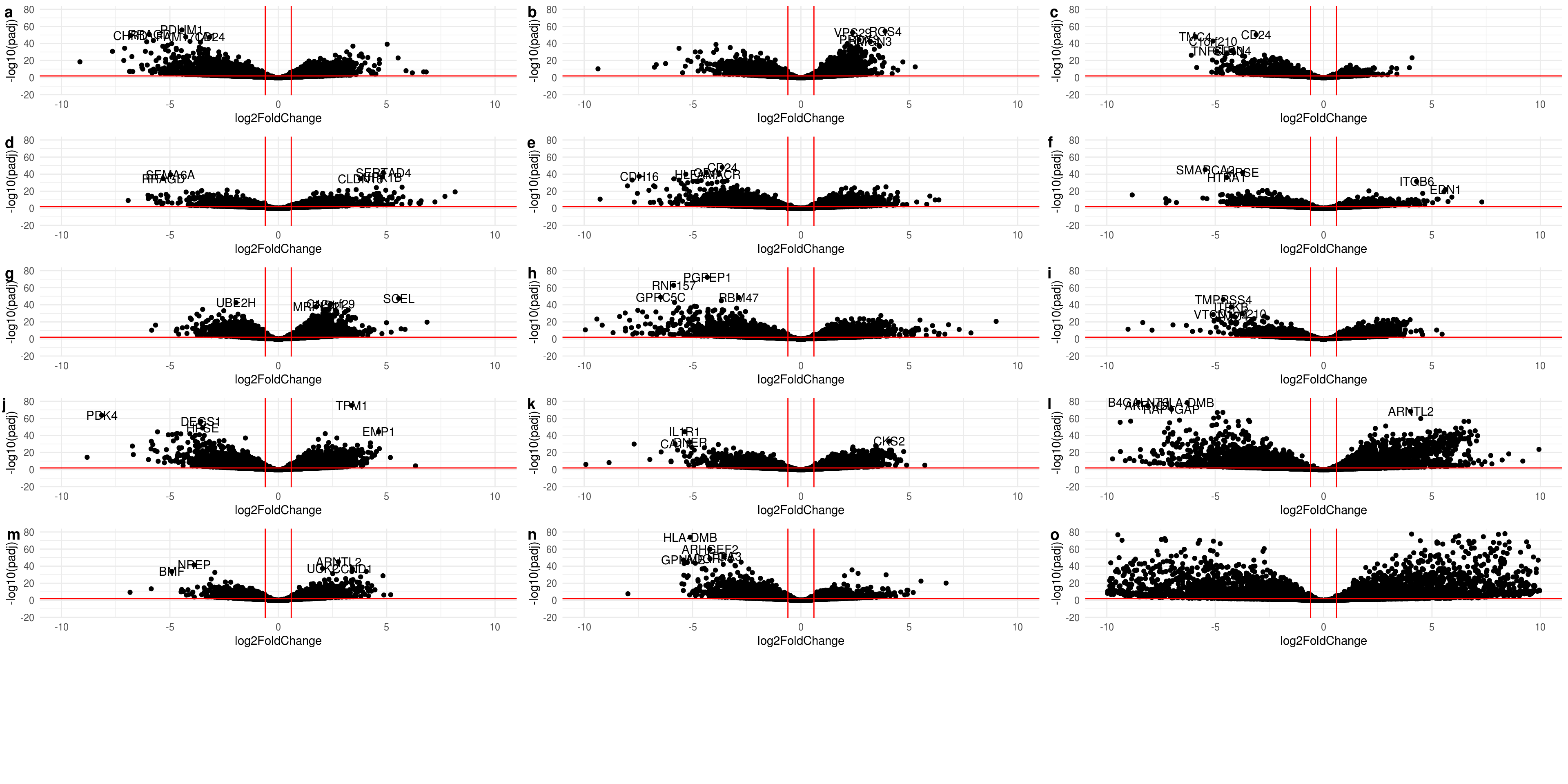
3.3.2.1 DE Gene / Cell Line Summary Table
summary.df <- do.call(cbind,lapply(res.ashr.list,function(x){data.frame(x) %>% dplyr::filter(padj<alpha & abs(log2FoldChange) > threshold) %>% dplyr::select(log2FoldChange) %>% summary %>% str_extract_all("-?[0-9]+.[0-9]+") %>% as.numeric}))
colnames(summary.df) <- ddsgrp@colData %>% as.data.frame %>% dplyr::select(cell.line) %>% unique %>% slice(-12) %>% mutate_all(as.character) %>% unname %>% unlist
rownames(summary.df) <- data.frame(res.ashr.list[[1]]) %>% dplyr::filter(padj<alpha & log2FoldChange > threshold ) %>% dplyr::select(log2FoldChange) %>% summary %>% str_extract_all("[1-9][:alpha:]+ [:alpha:]+|[:alpha:]+", simplify = TRUE)
summary.df <- rbind(summary.df, Count=sapply(res.ashr.list,function(x){data.frame(x) %>% dplyr::filter(padj<alpha & abs(log2FoldChange) > threshold) %>% dim %>% head(1) %>% as.integer() }) )
kable(summary.df)%>% kable_styling("striped") %>%scroll_box(width = "100%")| C5X | ES2 | HAC2 | HCH1 | JHOC5 | JHOC9 | KOC | OV207 | OVAS | OVISE | OVMANA | OVTOKO | SMOV2 | TOV21G | RMGII | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Min | -9.1504 | -10.53484 | -17.290 | -30.38630 | -30.111 | -24.6739 | -35.3516 | -18.0418 | -9.0214 | -17.0668 | -12.6053 | -24.5268 | -24.2218 | -30.9326 | -13.519 |
| 1st Qu | -2.2497 | -1.46009 | -1.915 | -1.80343 | -2.138 | -1.5273 | -1.3680 | -1.9656 | -1.7049 | -1.7811 | -1.7901 | -2.4433 | -1.3779 | -1.7976 | -1.919 |
| Median | -1.3674 | 1.00399 | -1.364 | -1.00537 | -1.252 | -1.0920 | 1.0488 | -1.1454 | -1.1332 | -1.1312 | -1.1083 | -1.2426 | 1.1312 | -1.1674 | 1.128 |
| Mean | -0.8333 | -0.01715 | -1.023 | 0.02051 | -0.666 | -0.2558 | 0.1445 | -0.4408 | -0.3192 | -0.2912 | -0.2817 | -0.5524 | 0.3168 | -0.4974 | 0.479 |
| 3rd Qu | 1.2309 | 1.49547 | 1.005 | 1.91022 | 1.313 | 1.4367 | 1.5807 | 1.4864 | 1.4829 | 1.4818 | 1.4657 | 1.5309 | 1.7226 | 1.3387 | 2.799 |
| Max | 6.8254 | 5.27310 | 12.381 | 8.15982 | 6.363 | 7.2999 | 6.8632 | 24.7988 | 5.4680 | 14.5713 | 5.7157 | 28.9316 | 41.5569 | 6.6958 | 25.399 |
| Count | 3532.0000 | 3170.00000 | 1519.000 | 1873.00000 | 2992.000 | 2614.0000 | 3135.0000 | 3451.0000 | 1701.0000 | 3664.0000 | 2898.0000 | 5136.0000 | 1902.0000 | 2391.0000 | 6434.000 |
56 DE genes is found across 16 cell line pairs
DGE.gene.list <- lapply(res.ashr.list,function(x){x %>% as.data.frame %>% dplyr::filter(padj<alpha & abs(log2FoldChange) > threshold) %>% rownames })
DGE.gene.union.list <- Reduce(union,DGE.gene.list) %>% unique
mtx<- sapply(DGE.gene.list,function(x){tmp<-c();tmp[DGE.gene.union.list %in% x] <- 1 ;return(tmp)})
colnames(mtx) <- ddsgrp@colData %>% as.data.frame %>% dplyr::select(cell.line) %>% unique %>% slice(-12) %>% mutate_all(as.character) %>% unname %>% unlist
rownames(mtx) <- DGE.gene.union.list
mtx[is.na(mtx)] <- 0
upset(data.frame(mtx),nset=15,order.by ="freq" ,point.size = 2, line.size = 1,mainbar.y.label = "DE Genes Overlap #", sets.x.label = " Total DE Genes")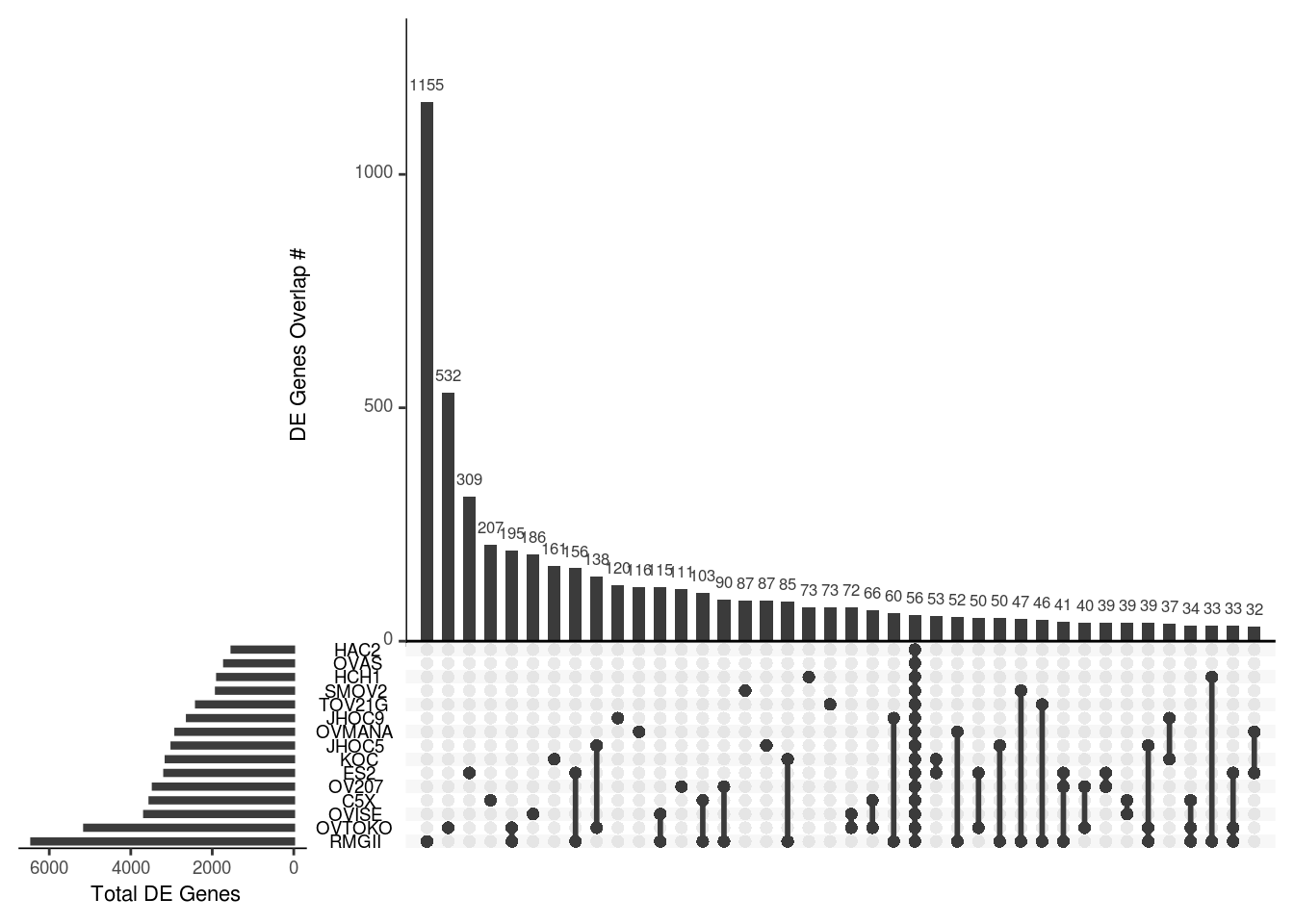
Fold change summary of 56 common DE genes
de.group.select.gene <- rownames(mtx[rowSums(mtx) == max(rowSums(mtx)),])
de.group.df <- do.call( "cbind" ,sapply(res.ashr.list, function(x){x %>% as.data.frame %>%rownames_to_column("gene") %>% dplyr::filter( gene %in% de.group.select.gene) %>% mutate_if(is.numeric,round,4) %>% dplyr::select(log2FoldChange)} ))
colnames(de.group.df) <- colnames(mtx)
rownames(de.group.df) <- de.group.select.gene
DT::datatable(de.group.df,extensions =c('Buttons', 'Scroller'), options = list(
dom = 'Bfrtip',
buttons = c('copy', 'csv', 'excel', 'pdf', 'print'),
"scrollX"= TRUE
))root.dir <- rprojroot::find_rstudio_root_file()
knitr::opts_chunk$set(echo = TRUE)
knitr::opts_knit$set(root.dir = root.dir )
library(readr)
library(tidyverse)
library(kableExtra)
library(stringr)
library(DiffBind)
source(file.path(root.dir,'src/util.R'))
source(file.path(root.dir,'src/plot_swoosh.R'))